

Sodium Adsorption, Diffusion and Coverage on Two-Dimensional GeP 3 Serdal Gurses, Fatih Ersan*
Department of Physics, Aydin Adnan Menderes University, Aydın, Turkey
*Corresponding author: Fatih Ersan
Citation: Gurses S and Ersan F (2020) Sodium Adsorption, Diffusion and Coverage on Two-dimensional GeP3. Sci Academique 1(1): 31-39.
Received date: 25 November, 2020; Accepted date: 14 December, 2020; Publication date: 19 December, 2020
Abstract
In this study, we performed density functional theory calculations to investigate Na adsorption and diffusion properties on two-dimensional germanium phosphide (III) (GeP3) monolayer. We found that single Na atom can strongly be adsorbed (2.71 eV) on the top of the valley site Ge atom and leads to a semiconductor to metal transition in GeP3. The adsorption energy of Na decreases by increased Na concentration. With full Na concentration, the GeP3 monolayer is subject to an increase in lattice parameter of about 0.25%. This increase rate is very small as desired in ion batteries. Moreover, Na atoms have very small diffusion barrier energy (0.25 eV) on GeP3 monolayers and the calculated diffusion coefficient at room temperature is 1.15×10−7 cm2/s. In addition, calculated average open-circuit voltage is 1.48 eV, which is comparable with the other Na-ion anode materials. Our results show that GeP3 monolayer can be a good candidate anode material for Na-ion batteries with a maximum capacity of 324 mAhg−1.
Introduction
Increasing demand for clean energy has encouraged researchers to develop novel energy storage devices, such as Lithium-ion (Li-ion) batteries. There are numerous experimental and theoretical studies on Li-ion batteries that focus on using two- dimensional (2D) materials as electrodes [1-6]. Several studies have revealed that graphene can store more Li atoms than graphite due to the former’s increased volume-surface ratio [7-9]. However, experimental studies show that transition metal dichalcogenides (TMDs) as MoS2 have higher Li capacities than graphene [10]. Ersan, et al. predicted that monolayer TMDs MX2 (M=Mo, W and X=O, S, Se, and Te) have lower activation energy barriers than graphene or silicene [11]. These lower energy barrier values result in higher diffusion coefficient for Li atoms, so that mobility of Li atoms on MX2 monolayers can be higher than many commercial anode materials. In addition, many theoretical studies focus on lithium adsorption on 2D semiconductor materials to investigate the electronic structure transition from semiconductor to metal [12,13].
However, limited Li resources necessitate new elements that can be used in energy storage. Since sodium (Na) and Li are in the same group in the Periodic table, they have similar properties. Hence, Na can be an excellent candidate for future large-scale energy storage applications. As mentioned above, graphene is suitable anode material for Li-ion battery applications with relatively high Li storage capacity [14]. In contrast, Malyi et al. showed that ideal graphene cannot be used as an anode for Na- ion batteries [15]. However, synthesized antimony/multilayer graphene heterostructure shows a high reversible Na storage capacity with a value of 452 mAhg−1 [16]. In another study, the synthesization of antimony carbon nanocomposite alloy was reported by Qian and co-workers [17]. It was found that this material has a long-term cycling stability with 94% capacity retention over 100 cycles, and it has Na storage capacity of 610 mAhg−1. By performing first-principles Density Functional Theory (DFT) simulations, Sengupta showed that 2D AlN can form a good Na-ion battery with a 385 mAhg−1 Na storage capacity, 1.86 V open-circuit voltage (OCV), and 0.15 eV diffusion barrier [18]. Similar to AlN (1.49 V), BN (1.46 V) has suitable cell voltage for Na-ion batteries and these cell voltages are comparable to carbon nanotubes [19]. Theoretical studies have shown that the maximum Na storage capacity of layered MoS2 is 146 mAhg−1, which is low than its Li storage capacity [20]. Increasing of Na coverage on MoS2 induces a phase transformation from 1H-MoS2 to 1T-MoS2.
Recently, a new 2D material GeP3 has been proposed by Heine et al [21]. The bulk form of GeP3 is metallic and has layered structure. Also, the calculated cleavage energy for monolayer GeP3 is 1.14 Jm−2 [21]. Adsorption and diffusion of Li atom on this newly predicted monolayer has been investigated by Zhang et al., and Li capacity has been reported as 648 mAhg−1 [22]. In addition, 2D GeP3 is found also can be good material as a high capacity anode material for non-Lithium-ion batteries [23]. Furthermore, it has also be pointed out that this monolayer can be a good candidate for gas sensor applications such as CH4, N2, and H2 molecules [24]. InP3 monolayer, which has the same structure as GeP3, is a promising anode material for both Li- and Na-ion batteries with a high storage capacity of 258 mAhg−1 [25].
In this study, we investigated Na adsorption and diffusion on 2D GeP3 monolayers by performing first-principles DFT calculations. Although Li and Na are in the same column of the Periodic table and accordingly share similar principles and properties, investigations have been focused mostly on Li-ion batteries instead of Na-ion batteries. Besides, Na resources are almost unlimited in the Earth’s crust and ocean, so, Na-ion batteries are more economical than Li-ion batteries. Therefore, exploration of the nature of Na-ion batteries will be very important to improving energy storage applications.
Computational Details
In this study, first-principles DFT is used to investigate adsorption and diffusion properties of a Na atom on the surface of a GeP3 monolayer. The calculations are performed using spin-polarized, plane-wave DFT with ultrasoft pseudopotentials [26] as implemented in Quantum ESPRESSO software package [27]. For the exchange-correlation functional, generalized gradient approximation (GGA) is employed by using Perdew-Burke-Ernzerhof (PBE) parametrization [28]. The van der Waals (vdW) corrections are taken into account by using DFT-D2 [29]. For the self-consistent field potential and total energy calculations, the Brillouin Zone (BZ) is sampled using a (12×12×1) Monkhorst-Pack special k-points mesh for monolayer GeP3 unit-cell [30]. A plane-wave basis set with a kinetic energy cutoff of 70 Ry is used for all calculations. The lattice constants and atomic positions are optimized by using the BFGS quasi-Newton algorithm [31]. The convergence criterion for energy is chosen as 10−6 Ry between two successive iteration steps, and the maximum Hellmann-Feynman forces exerted on each atom is reduced to less than 0.005 Ry/Å upon ionic relaxation. To avoid interlayer interactions, two GeP3 monolayers are separated by a distance of 20Å vacuum length. We used the climbing image nudged elastic band (CI-NEB) method to seek the minimum energy path and saddle points between determined initial and final positions [32].
In order to determine the adsorption energy Eads of Na atoms on a GeP3 monolayer we used the following equation;

where ENa and EGeP3 are the total energy of the isolated Na atom and the total energy of bare GeP3 monolayer, respectively. EGeP3+Na is the total energy of the Na atom adsorbed EGeP3 monolayer.
Results and Discussion
For the sake of comparison, we first carried out the properties of a monolayer structure of GeP3. The calculated optimized lattice constants for the 2×2 supercell are a=b=13.87Å. We also evaluated the cohesive energy for GeP3 monolayer by Ecoh = (mEGe + nEP −EGeP3)/(m + n), where EGeP3 is the total energy of the bare monolayer, and EGe and EP are the total energies of isolated free atoms of Ge and P, respectively. m and n are the numbers of Ge and P atoms in the unit cell. The calculated cohesive energy is 3.43 eV/atom. Figure 1 depicts the atomic structure, band structure, and total and partial electronic density of states of the monolayer GeP3. The PBE calculation predicted that the band gap of the GeP3 monolayer is 0.22 eV and the band character is indirect from
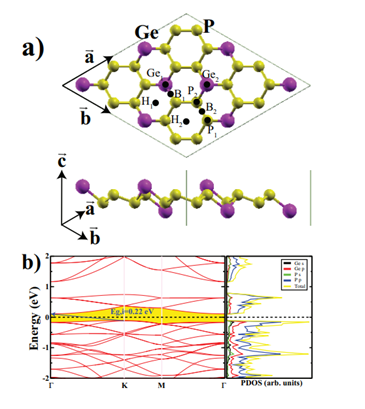
Figure 1: (Color online) a) Top and side views of the GeP3 monolayer, eight considered adsorption sites on the GeP3 monolayer are also shown. b) The electronic band structure and corresponding total and partial density of states of the 2×2 supercell of GeP3 monolayer. The Fermi level is set to zero energy. Indirect band character is shown by a blue arrow.
The K (valence band maximum; VBM) to the Γ (conduction band minimum; CBM). Both the CBM and VBM are occupied dominantly by phosphorus p orbitals; the second highest contribution comes from germanium p orbitals. These calculated values are consistent with the previous results [21,22,24].
We next investigated the adsorption of a single Na atom on the GeP3 monolayer. We used (2×2) supercells to prevent the Na-Na interactions between the neighbor cells. With this enlargement, the corresponding chemical stoichiometry is Ge8P24Na. As illustrated in Figure 1(a), eight adsorption sites were considered to determine the most energetically favorable positions for a single Na atom bound to GeP3. These sites are: the top of Ge atoms (Ge1 is for valley site Ge atoms, Ge2 is for the top of upper-level Ge atoms), top of P atoms (P1, P2), top of hollow sites (H1, H2) and top of the bridge sites (B1, B2). Our calculations show that the most favorable adsorption site for Na atom is Ge1 with a Eads value of 2.71 eV. The most favorable adsorption sites from best to worst are as follows: Ge1=P1 > H1 = H2 > P2 > Ge2. The corresponding adsorption energies are 2.71 eV > 2.65 eV > 2.60 eV > 2.02 eV. During geometrical optimization, a Na atom on a P1 site slips towards a Ge1 site, and a Na atom on a H1 site also slips close to a Ge1 site. These adsorption energies prove that there is a strong interaction between the Na atom and the GeP3 monolayer.
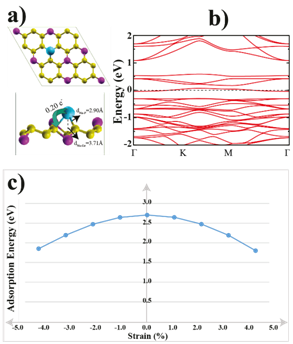
Figure 2: (Color online) a) Top and side views of the single Na atom adsorbed GeP3 monolayer with bond distances. The arrow shows the electron transfers from Na atom to the surface. b) The electronic band structure of the Na adsorbed GeP3 system. c) Adsorption energies of a single Na atom as the function of applied biaxial tensile/compressive strain percentage.
Figure 2(a) and (b) illustrate the atomic structure of the Na atom adsorbed GeP3 monolayer with the bond distances and electronic band dispersion after the geometric optimization. Löwdin analysis indicates that 0.20 electrons transferred from Na atom to the GeP3 monolayer while Na atom on Ge1 site. This amount of charge transfer results in the shifting of the Fermi energy level of the GeP3 and semiconducting-metal transition occurs and eventually, the new structure becomes a non-magnetic metal which has a lower resistivity and higher electronic conductivity than bare GeP3 (see Figure 2(b)). The lattice constants are decreased only 0.14% by single Na atom adsorption and this extremely small volume change during Na intercalation may solve the volume expansion issue in Na-ion batteries. To see how the applied tensile or compressive strain affect the binding energy of Na atom, we applied biaxial in-plane strains to the monolayer GeP3. From Figure 2(c), we can see that the adsorption energy decreases quickly from 2.71 eV to 1.80 eV with tensile or compressive strains. This can imply that Na atom adsorption energy is sensitive to the external strain.
Even though low electrical resistivity is important to determine the rate performance of anode materials, it is not sufficient by itself. The adsorbed atom diffusion performance should also be known. Therefore, the diffusion of Na atom on GeP3 monolayer is also examined in this study. To seek the minimum energy path and saddle points, we use the climbing image nudged elastic band (CI-NEB) method [32]. For these calculations,
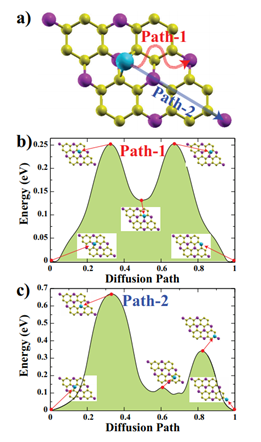
Figure 3: (Color online) a) Top view of the considered diffusion paths for Na (Path-1 (red arrow), Path-2 (blue arrow)). b) Calculated energy profiles along the Path-1 and c) Path-2 for one Na atom on the GeP3 monolayer. Insets depict the atomic structures for the transition states in Path-1 and Path-2.
We determined two linear different paths to determine the activation barrier energy of the Na atom. Besides, the selected initial and final images are fully optimized before the CI-NEB calculation. All diffusion paths divided by 23 equal intervals, at the end of the CI-NEB calculations the paths that Na atom follows can see in Figure 3(a) with red and blue arrows.
For both Path-1 and Path-2, there are two energy minima, one of them is absolute energy minimum where the Na atom is adsorbed on Ge1 site, and the other one is a local minimum corresponding to the Na atom close to H2 site. The two maxima occur while Na atom is passing from the B1 sites for the Path-1. Due to Ge2 site is the least favorable adsorption site among the all considered sites, high energy barrier happens when the Na atom is on this site (see inset of Figure 2(c)). The calculated activation energy barriers for these two paths for Na atom are 0.25 eV and 0.66 eV for Path-1 and Path-2, respectively. These energy barriers are comparable with Na atom on InP3 monolayer which is obtained by Liu, et al. [25] From these CI-NEB calculations we can easily say that Na atom prefers to diffuse on the line of Path-1.
The diffusion coefficient (D) at the room temperature of the Na atom on GeP3 can be calculated using Arrhenius equation by the following equation:

where a is the lattice constant of GeP3 monolayer (a=6.935Å), v=1×1011 Hz (jumping frequency), and kBT =0.026 eV. The calculated diffusion coefficient along the Path-1 is 1.15×10−7 cm2/s and this D value indicates that Na atom can easily move on the GeP3 which is important for the electrochemical reactions in ion batteries.
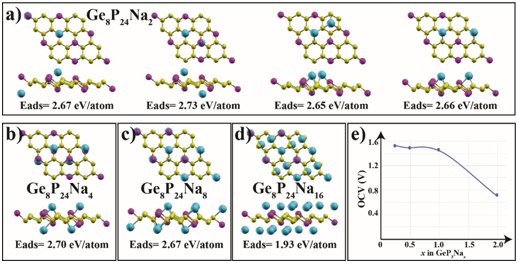
Figure 4: (Color online) a) Top and side views of the optimized atomic structures of Ge8P24Na2 (GeP3Na0.25), b) Ge8P24Na4 (GeP3Na0.5), c) Ge8P24Na8 (GeP3Na), and d) Ge8P24Na16 (GeP3Na2), e) The electrode potential profiles of Na interactions on GeP3.
As is known, the performance of the Na(Li)-ion batteries is directly related to the storage capacity. For this reason, we further examined more Na atoms adsorbed on both sides of the GeP3 monolayer to specify the highest theoretical Na atom-specific capacity. For this investigation, we first considered two individual Na atoms in two main scenarios. In the first scenario, the Na atoms are adsorbed to different sides of the same GeP3 monolayer. In the other case, both Na atoms are adsorbed to the same side. Both cases are illustrated in Figure 4(a). Our calculations show that the adsorption energy of the Na atoms on opposite sides is higher by 60 meV/atom than that of the Na atoms on one side. This implies that two Na atoms prefer to bind GeP3 separately on both sides to counteract the possible structural distortion in GeP3. Figure 4(b), (c), and (d) show the atomic structures and adsorption energies for the increased number of Na atoms on the GeP3 monolayer. After full coverage of the Na atoms on the Ge1 sites (see Figure 4(c)), we placed eight more Na atoms on P2 sites on top of the Na atoms level. After geometrical optimization, these additional Na atoms penetrated and remained between the lower level Na atoms without creating Na clusters (see Figure 4(d)). Thus all Na atoms were positioned on the same level for each side of the GeP3 monolayer.
The maximum theoretical capacities (C) are estimated by the following equation:

where MGeP3 is the atomic mass of the GeP3 monolayer, x is the highest number of adsorbed Na atoms, and F is the Faraday constant. The calculated maximum Na storage capacity for the GeP3 monolayer is 324 mAhg−1 which is higher than that of InP3 monolayer (258 mAhg−1) [25], because of the relatively light atomic weight of the Ge atom. In addition, this Na storage capacity on GeP3 is larger than those on many transition metal tellurides (MTe2; M=Co, Fe, Mn, Sc, Ti : 22-130 mAhg−1)[33], amorphous (Glassy) carbon (173 mAhg−1)[34] and MoS2 (146 mAhg−1)[35]. From these results, we can say that if the GeP3 monolayer structure is used as an anode material for Na ion batteries, it could show high power density performance. Also, the full coverage of Na atoms on GeP3 results in just a 0.25% lattice constant enhancement for a 2×2 supercell of GeP3, which is a negligible change in the volume of the cell.
Disregarding entropic and enthalpic energy contributions, the average open-circuit voltage (VOCV) can be calculated from total-energy differences with the following equation:
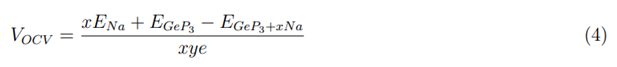
where EGeP3+xNa and EGeP3 are the total energies of GeP3 with and without Na intercalation. ENa is the cohesive energy of metal Na; x is the number of adsorbed Na atom on the GeP3 and y is the electrical charge of Na ions in the electrolyte (we assume that y = 1 in this equation). Figure 4(e) shows the calculated average open-circuit voltage profile of Na on GeP3. As the Na atom concentration increases from 0.25 to 2.0, the electrode potential (U) tends to decrease. However, we should note that the biggest decrease occurs between GeP3Na2 to GeP3Na, while for other concentrations the potential alteration is only 0.05 V. The calculated average VOCV for GeP3Na0.25 to GeP3Na is 1.48 V, which is similar to the commercial anode material of TiO2 (1.50V) [36].
Conclusion
In conclusion, we systematically investigated the adsorption and diffusion of sodium on the GeP3 monolayer using first-principles density functional calculations. We also studied the external strain effects on a single Na atom adsorbed on a GeP3 monolayer, and found that adsorption energy of Na atom on GeP3 is reduced by tensile or compressive strain. The semiconducting GeP3 monolayer turns to metal upon adsorption of a single Na atom, which leads to an increase of electrical conductivity of the material. With full Na concentration, the GeP3 monolayer undergoes only a 0.25% lattice parameter increase and the theoretical Na storage capacity is 324 mAhg−1. Besides, the calculated average VOCV from GeP3Na0.25 to GeP3Na is 1.48 V, and is comparable to the electrode potential of the commercial anode materials. Our results indicate that the GeP3 monolayer is a promising anode material for Na-ion battery applications similar to other newly predicted two-dimensional materials [37,42].
Acknowledgments
Computing resources used in this work were provided by the TUBITAK ULAKBIM, High Performance and Grid Computing Center (Tr-Grid e-Infrastructure).
References
- Theerthagiri J, Senthil RA, Senthilkumar B, Polu AR, Madhavan J, et al. (2017) Recent advances in MoS2 nanostructured materials for energy and environmental applications–a review. Journal of Solid State Chemistry 252: 43-71.
- Lei Y, Fujisawa K, Zhang F, Briggs N, Aref AR, et al. (2019) Synthesis of V-MoS2 Layered Alloys as Stable Li-Ion Battery Anodes. ACS Applied Energy Materials 2: 8625-32.
- Rojaee R, Shahbazian-Yassar R (2020) Two Dimensional Materials to Address the Li-Based Battery Challenges. ACS Nano 3: 2628-2658.
- Sarikurt S (2019) A First-Principles Investigation of Lithium Adsorption and Diffusion on BN, AlN and GaN Monolayers. Eski¸sehir Tech. Univ. J. of Sci. and Technology A –Appl. Sci. and Eng 20: 436-445.
- Ersan F, Gokoglu G, Akturk E (2015) Adsorption and Diffusion of Lithium on Monolayer Transition Metal Dichalcogenides (MoS2(1–x)Se2x) Alloys, The Journal of Physical Chemistry C 119: 28648-28653.
- Akgenc B (2019) Two-dimensional black arsenic for Li-ion battery applications: a DFT study, Journal of Materials Science 54: 9543-9552.
- Tang Q, Zhou Z, Chen Z (2013) Graphene-related Nanomaterials: Tuning Properties by Functionalization. Nanoscale 5: 4541-4583.
- Medeiros PVC, Brito Mota F de, Mascarenhas AJS, Castilho CMC de (2010) Adsorption of Monovalent Metal Atoms on Graphene: A Theoretical Approach. Nanotechnology 21: 115701.
- Yang CK (2009) A Metallic Graphene Layer Absorbed with Lithium. Appl. Phys. Lett 94: 163115.
- Hwang H, Kim H, Cho J (2011) MoS2 Nanoplates Consisting of Disordered Graphene-like Layers for High Rate Lithium Battery Anode Materials. Nano Lett 11: 48264830.
- Ersan F, Ozaydin HD, Gokoglu G, Akturk E (2017) Theoretical investigation of lithium adsorption, diffusion and coverage on MX2 (M = Mo, W; X = O, S, Se, Te) monolayers. Applied Surface Science 425: 301-306.
- Gokce A, Ersan F (2017) Adsorption of alkali and alkaline earth metal atoms and dimers on monolayer germanium carbide, Philosophical Magazine 97: 155-167.
- Kadioglu Y, Ersan F, Gokoglu G, Akturk OU, Akturk E (2016) Adsorption of alkali and alkaline- earth metal atoms on stanene: A first-principles study, Materials Chemistry and Physics180: 326-331.
- Garay-Tapia AM, Romero AH, Barone V (2012) Lithium Adsorption on Graphene: From Isolated Adatoms to Metallic Sheets. J. Chem. Theory Comput 8: 1064-1071.
- Malyia OI, Sopihac K, Kulishc VV, Tand TL, Manzhosb S, et al. (2015) A computational study of Na behavior on graphene. Applied Surface Science 333: 235-243.
- Hu L, Zhu X, Du Y, Li Y, Zhou X, et al. (2015) A Chemically Coupled Antimony/Multilayer Graphene Hybrid as a High-Performance Anode for Sodium-Ion Batteries. Chem. Mater 27: 8138-8145.
- Qian J, Chen Y, Wu L, Cao Y, et al. (2012) High capacity Na-storage and superior cyclability of nanocomposite Sb/C anode for Na-ion batteries. Chem Commun 48: 7070–7072.
- Sengupta A (2018) Lithium and sodium adsorption properties of two-dimensional aluminum nitride. Applied Surface Science 451: 141-147.
- Hosseinian A, Khosroshahi ES, Nejati K, Edjlali E, Vessally E (2017) A DFT study on graphene, SiC, BN, and AlN nanosheets as anodes in Na-ion batteries. J Mol Model 23: 354.
- Mortazavi M, Wang C, Deng J, Shenoy VB, Medhekar NV (2014) Ab initio characterization of layered MoS2 as anode for sodium-ion batteries. Journal of Power Sources 268: 279-286.
- Jing Y, Ma Y, Li Y, Heine T (2017) GeP3: A Small Indirect Band Gap 2D Crystal with High Carrier Mobility and Strong Interlayer Quantum Confinement. Nano Lett 17: 1833-1838.
- Zhang C, Jiao Y, He T, Ma F, Kou L, et al. (2017) Two-dimensional GeP3 as a high capacity electrode material for Li-ion batteries. Phys. Chem. Chem. Phys 19: 25886-25890
- Deng X, Chen X, Huang Y, Xiao B, Du H (2019) Two-Dimensional GeP3 as a High Capacity Anode Material for Non-Lithium-Ion Batteries. J. Phys. Chem C 8: 4721–4728
- Tian B, Huang T, Guo J, Shu H, Wang Y, et al. (2019) Gas adsorption on the pristine monolayer GeP3: A first-principles calculation. Vacuum 164: 181-185.
- Liu J, Liu CS, Ye XJ, Yan XH (2018) Monolayer InP3 as a reversible anode material for ultrafast charging lithium- and sodium-ion batteries: a theoretical study. J. Mater. Chem A 6: 3634.
- Quantum ESPRESSO pseudopotentials.
- Giannozzi P, Baroni S, Bonini N, Calandra M, Car R, et al. (2009) Quantum ESPRESSO: a modular and open-source software project for quantum simulations of materials. J Phys Condens Matter 21: 395502.
- Perdew JP, Burke K, Ernzerhof M (1996) Generalized Gradient Approximation Made Simple, Phys. Rev. Lett 77: 3865.
- Grimme S (2006) Semiempirical GGA-type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem 27: 1787-1799.
- Monkhorst HJ, Pack JD (1976) Special points for Brillouin-zone integrations. Phys. Rev. B 13: 5188.
- Broyden CG (1969) A new double-rank minimization algorithm. Notices Amer. Math. Soc 16: 670
- Henkelman G, Jonsson H (2000) A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys 113: 9901-9904.
- Yi-Xuang Li, Putungan DB, Lin SH (2018) Two-dimensional MTe2 (M = Co, Fe, Mn, Sc, Ti) transition metal tellurides as sodium ion battery anode materials: Density functional theory calculations. Physics Letters A 382: 2781–2786.
- Legrain F, Sottmann J, Kotsis K, Gorantla S, Sartori S, et al. (2015) Amorphous (Glassy) Carbon, a Promising Material for Sodium Ion Battery Anodes: a Combined First-Principles and Experimental Study. J. Phys. Chem. C 119: 1349613501.
- Mortazavi M, Wang C, Deng J, Shenoy VB, Medhekar NV (2014) Ab initio characterization of layered MoS2 as anode for sodium-ion batteries. J. Power Sources 268: 279–286.
- Yang Z, Choi D, Kerisit S, Rosso KM, Wang D, et al. (2009) Nanostructures and lithium electrochemical reactivity of lithium titanites and titanium oxides. J. Power Sources 192: 588–598.
- Luo W, Shen F, Bommier C, Zhu H, X. Ji L. Hu, (2016) Na-ion battery anodes: materials and electrochemistry. Accounts of chemical research 49: 231-240.
- Bhuvaneswari R, Nagarajan V, Chandiramouli R (2020) Investigation on bare and hydrogenated Sb-nanosheets as an electrode material for Na-ion battery-A DFT study. Physica B: Condensed Matter 562: 75-81.
- Moalla A, Noei M, Khazali F, Maleki A (2020) A computational study on the BN-yne sheet application in the Na-ion batteries, Journal of Molecular Graphics and Modelling 97: 107567.
- Wei X, Ma M, Zhang H, Wang L, Dong C (2021) Density functional theory study of ultra-thin InSe electrodes for Na and Mg ion storage and transport. Materials Letters 285: 129091.
- Peyghan AA, Beheshtian J (2020) Application of hexa‐peri‐hexabenzocoronene nanographene and its B, N, and Bn doped forms in Na-ion batteries: A density functional theory study, Thin Solid Films 704: 137979.
- Guo Y, Bo T, Wu Y, Zhang J, Lu Z, et al. (2020) YS2 monolayer as a high-efficient anode material for rechargeable Li-ion and Na-ion batteries. Solid State Ionics 345: 115187.