

Quantum Computational, Spectroscopy Investigation (FTIR, FT-Raman), HOMO-LUMO and Docking Studies on Selegiline B. Aysha Rifana1,2, Shyam Sundar1, Johanan Christian Prasana1,2*, A. Anuradha2,3
1Department of Physics, Madras Christian College, East Tambaram, Chennai 600059, Tamil Nadu, India
2University of Madras, Chennai, 600005, Tamil Nadu, India
3PG & Research Department of Physics, Queen Mary’s College, Chennai 600004, Tamil Nadu, India
*Correspondence to: Johanan Christian Prasana
Citation: Rifana BA, Sundar S, Prasana JC, Anuradha A (2022) Quantum Computational, Spectroscopy Investigation (FTIR, FT-Raman), HOMO-LUMO and Docking Studies on Selegiline. Sci Academique 3(2): 44-49
Received: 20 September, 2022; Accepted: 04 October 2022; Publication: 08 October 2022
Abstract
Computational analysis has been a powerful tool in characterizing a compound. With its high precision, accurate results can be obtained for any given compound. Density Functional Theory, a quantum mechanical computational method was used to study vibrational spectra of the title compound. Relative and absolute values of FTIR and FT-Raman were found. Potential energy distribution percentage for each vibrational mode was calculated. The calculated HOMO and LUMO energies were -6.1022 eV and -0.4493 eV, respectively, resulting in a band gap energy of 5.6529 eV, indicating charge transfer within the molecule. UV spectra of the title compound were observed by TD-DFT method. Electron localization function (ELF), Localized orbital locator (LOL), Molecular electrostatic potential (MEP) of the title compound were also obtained. Drug likeness values were analyzed to assess the title compound’s potential as an active pharmaceutical component. Biological nature of the compound was observed by molecular docking studies.
Introduction
Monoamine oxidase (MAO), discovered by Balschko in the 1930s, is one of the most important enzymes in neurotransmitter metabolism [1]. MAO has the potential to be used in the treatment of several neurodegenerative diseases, including Parkinson’s and Alzheimer’s. MAO-catalyzed reactions generate hydrogen peroxide, a source of hydroxyl radicals, and MAO inhibitors may thus be useful in managing the outcome of stroke and other tissue damage caused by oxidative stress [2]. Selegiline (title compound) was developed in Hungary by Knoll et al. in 1964 as a novel monoamine oxidase (MAO)-inhibitor antidepressant. In patients with early Parkinson’s disease (PD), Selegiline postpones the need for levodopa therapy [3]. From the deprenyl and tocopherol antioxidative treatment, selegiline, with or without tocopherol, reduces physical and psychological deficits in patients with PD within first month of treatment and reduces the probability of reaching a primary endpoint, the decision to treat with levodopa [4]. The empirical formula of selegiline is C13H17N, and IUPAC name is ethyl (2R)-N-methyl-1-Phenyl-N-prop-2-ynylpropan-2-amine.
According to literature survey, a few spectroscopic and in vitro assays have already been published on the title compound [5,6]. The present study provides a complete vibrational, electronic and topological analysis under theoretical background. This paper describes a comprehensive spectroscopic investigation of the title compound at the B3LYP/6-311++G (d, p) level of theory. Frontier molecular orbitals (HOMO, LUMO) determine how the molecule interacts with other species, allowing us to characterize the chemical reactivity of the molecule. MEP (Molecular Electrostatic potential), ELF (Electron localization function) and LOL (Localized orbital locator) were used to examine the distribution of electrons and reactive sites on the surface of the title compound. Drug likeness were also carried out. Molecular docking analysis is performed by selecting suitable protein targets to study the bio activeness of the title compound.
Computational Details
The molecular parameters of the title compound in ground state were calculated using DFT B3LYP/6-311++G (d, p) basis set. In DFT methods, Becke’s three(B3) combined with Lee, Yang and Parr(LYP) method is the best predicting results for molecular geometry and vibrational wave numbers for moderately larger molecule [7,8]. Chemcraft 1.8 [9] was used to visualize optimized geometrical structure of the title compound. Vibrational assignment calculations in terms of PED contributions were performed with high accuracy using VEDA software. [10]. Molecular electrostatic potential (MEP) and HOMO-LUMO studies were carried out using Gauss View 5.0software [11-13]. Localized Orbital Locator (LOL) and Electron Localization Function (ELF) two-dimensional plots were obtained using MULTIWFN 3.4.1 [14]. Swiss ADME Tool is used to determine the drug likeness nature and ADME properties of the title compound [15]. The binding energy, inhibition constant, and other biological parameters of ligand-protein interaction were determined using the AutoDock 4.2.1 programme [16].
Results and Discussion
Molecular Geometry
Bond parameters (bond angle and bond length) of the title compound were obtained by DFT/B3LYP method with basis set 6-311++G (d, p). The optimized geometrical structure is shown in Fig. 1. Bond angle, the angle between three atoms and bond length, the distance between two neighboring atoms values obtained were tabulated in table 1. Homonuclear atoms such as C12-C15, C15-C17 and C6-C12 have the highest bond lengths of 1.553 Å, 1.532 Å and 1.513 Å. On the other hand, heteronuclear atoms such as C15-N21, N21-C26 and N21-C22 have the highest bond lengths of 1.483 Å,1.470 Å and 1.463 Å. Least bond length values were found in atoms C30-H3, C3-H9 and C1-H7 of values 1.063,1.084 and 1.085. In the case of bond angle, the maximum values obtained were 121.3°, 121.1° and 121.1° which corresponded to the atoms C1-C6-C12, C4-C5-C6 and C2-C1-C6 respectively. Presence of benzene ring structure comprised of atoms C1 to C6 was observed in the title compound.
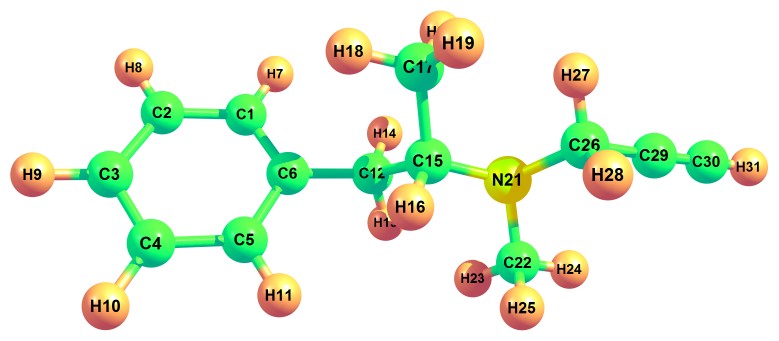
Figure 1: Optimised geometric structure of (2R)-N-methyl-1-Phenyl-N-prop-2-ynylpropan-2-amine.
| Bond Length (Å) | B3LYP/6-311++G (d, p) | Bond Angle (°) | B3LYP/6-311++G (d, p) |
| C1-C2 | 1.399 | C2-C1-C6 | 121.1 |
| C1-C6 | 1.399 | C2-C1-H7 | 119.6 |
| C1-H7 | 1.085 | C1-C2-C3 | 120.1 |
| C2-C3 | 1.393 | C1-C2-H8 | 119.8 |
| C2-H8 | 1.085 | C6-C1-H7 | 119.3 |
| C3-C4 | 1.395 | C1-C6-C5 | 118.1 |
| C3-H9 | 1.084 | C1-C6-C12 | 121.3 |
| C4-C5 | 1.393 | C3-C2-H8 | 120.1 |
| C4-H10 | 1.085 | C2-C3-C4 | 119.4 |
| C5-C6 | 1.401 | C2-C3-H9 | 120.3 |
| C5-H11 | 1.086 | C4-C3-H9 | 120.3 |
| C6-C12 | 1.513 | C3-C4-C5 | 120.2 |
| C12-H13 | 1.091 | C3-C4-H10 | 120.0 |
| C12-H14 | 1.094 | C5-C4-H10 | 119.8 |
| C12-C15 | 1.553 | C4-C5-C6 | 121.1 |
| C15-H16 | 1.106 | C4-C5-H11 | 119.4 |
| C15-C17 | 1.532 | C6-C5-H11 | 119.5 |
| C15-N21 | 1.483 | C5-C6-C12 | 120.6 |
| C17-H18 | 1.091 | C6-C12-H13 | 108.4 |
| C17-H19 | 1.091 | C6-C12-H14 | 110.2 |
| C17-H20 | 1.095 | C6-C12-H15 | 114.3 |
| N21-C22 | 1.463 | C3-C12-14 | 106.4 |
| N21-C26 | 1.470 | H13-C12-C15 | 109.6 |
| C22-H23 | 1.089 | H14-C12-C15 | 107.8 |
| C22-H24 | 1.090 | C12-C15-H16 | 108.1 |
| C22-H25 | 1.106 | C12-C15-C17 | 109.2 |
| C26-H27 | 1.089 | C12-C15-N21 | 109.1 |
| C26-H28 | 1.108 | H16-C15-C17 | 107.6 |
| C26-C29 | 1.464 | H16-C15-N21 | 109.3 |
| C29-C30 | 1.202 | C17-C15-N21 | 113.4 |
| C30-H31 | 1.063 | C15-C17-H18 | 109.2 |
| C15-C17-H19 | 112.8 | ||
| C15-C17-H20 | 110.9 | ||
| C15-N21-C22 | 111.8 | ||
| C15-N21-C26 | 114.1 | ||
| H18-C17-H19 | 107.0 | ||
| H18-C17-H20 | 108.0 | ||
| H19-C17-H20 | 108.8 | ||
| C22-N21-C26 | 109.8 | ||
| N21-C22-H23 | 111.0 | ||
| N21-C22-H24 | 109.6 | ||
| N21-C22-H25 | 112.2 | ||
| N21-C26-H27 | 109.2 | ||
| N21-C26-H28 | 112.0 | ||
| N21-C26-C29 | 113.0 | ||
| H23-C22-H24 | 107.5 | ||
| H23-C22-H25 | 108.2 | ||
| H24-C22-H25 | 108.3 | ||
| H27-C26-H28 | 106.8 | ||
| H27-C26-C29 | 107.3 | ||
| H28-C26-C29 | 108.3 | ||
| C26-C29-C30 | 178.0 | ||
| C29-C30-H31 | 179.6 |
Table 1: Geometrical parameters Bond Length (Å) and Bond Angle (°) optimized in (2R)-N-methyl-1-Phenyl-N-prop-2-ynylpropan-2-amine with basis set 6-311++G (d, p).
Vibrational Analysis
Vibrational analysis of the title compound was carried out in detail through FT-IR and FT-RAMAN studies. For a non-linear compound, the title compound will have n = 3N-6 normal vibrational modes, i.e., n = 87. The contribution of vibrational frequency by a particular set towards the potential energy were represented as potential energy distribution (PED%) and their values are tabulated in table 2 [17]. A factor of 0.961 was multiplies to the unscaled frequency values to get the scaled values as they are more refined and accurate. Types of vibration such as stretching, bending and torsional were given by the vibrational assignment. All vibrational calculations were carried out by GAUSSIAN 09W with basis set, B3LYP/6-311++G (d, p). fig.2 and fig.3 represents the theoretical FT-IR and FT-RAMAN spectra.
C-H Vibration
Heteroaromatic organic compound and its derivatives commonly exhibit multiple peaks in the region 3100 to 3000 cm-1 [18,19]. The C-H stretching bands of the title compound was reported in the range 3340 to 2773 cm-1. The highest Stretching 98% was observed in the vibrational region of 2911 cm-1 and 2693 cm-1. Pure stretching was not found in any region.
C-C Vibration
The bonds between 1650 and 140 cm-1 range in the aromatic and heteroaromatic compounds are assigned to carbon-carbon vibration [20]. In the present study, Theoretical frequencies assigned to C-C stretching vibrations are 2131 cm-1,1591 cm-1 and maximum PED contribution to this vibration is 96%.
C-N Vibration
Mixing of several modes is possible in the region makes the documentation of C-N bonds very difficult. Frequency nearer to 1500 cm-1 indicates C=N bonds while frequency nearer to 1300 cm-1 indicates the presence of C-N bonds [21]. For the title compound, C-N stretching vibrations were found at 1201 ,1127 ,1041 ,974, 914 and 777 cm-1.
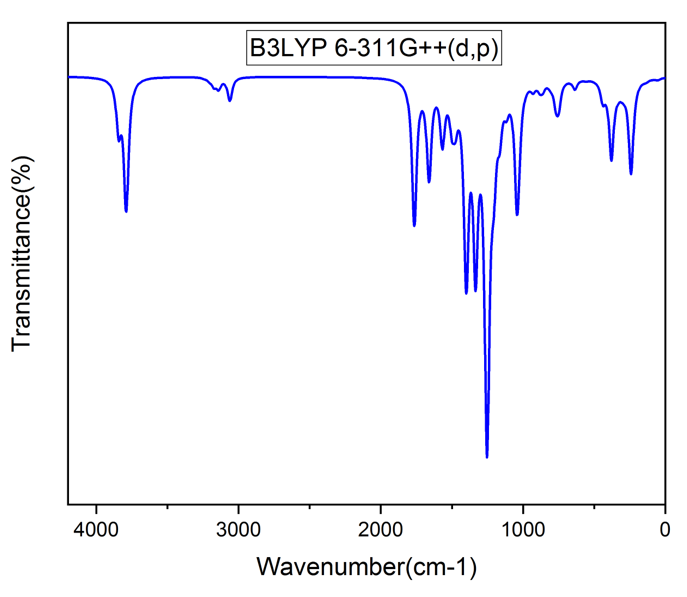
Figure 2: FT-IR Spectra.
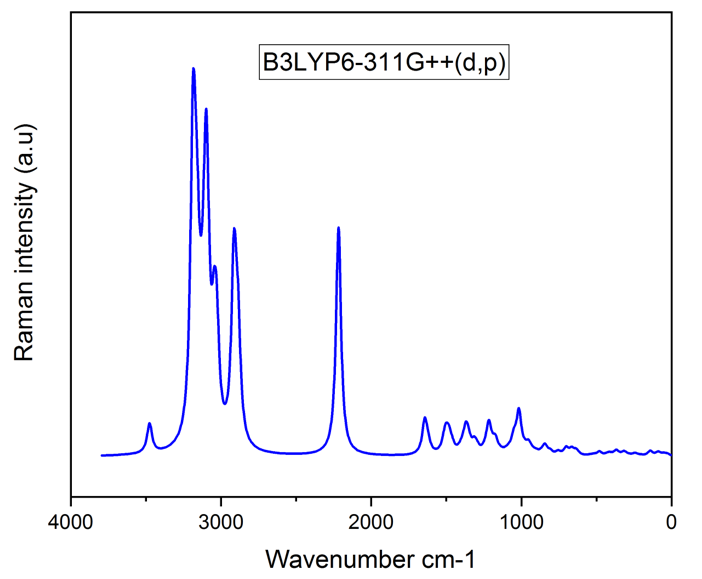
Figure 3: FT-RAMAN Spectra.
|
Modes |
CM-1 |
IR Intensity |
Raman Activity |
Vibrational Assignment PED% |
|||
|
Unscaled |
Scaled |
Relative |
Absolute |
Relative |
Absolute |
||
|
87 |
3476 |
3340 |
77 |
62 |
37 |
10 |
STRE CH(95) |
|
86 |
3187 |
3063 |
16 |
13 |
325 |
100 |
STRE CH(94) |
|
85 |
3176 |
3052 |
31 |
25 |
44 |
12 |
STRE CH(93) |
|
84 |
3167 |
3044 |
7 |
6 |
111 |
31 |
STRE CH(93) |
|
83 |
3156 |
3033 |
3 |
3 |
66 |
19 |
STRE CH(86) |
|
82 |
3152 |
3029 |
9 |
7 |
32 |
9 |
STRE CH(90) |
|
81 |
3129 |
3007 |
25 |
20 |
38 |
1 |
STRE CH(94) |
|
80 |
3112 |
2991 |
24 |
20 |
31 |
9 |
STRE CH(95) |
|
79 |
3104 |
2983 |
7 |
6 |
128 |
36 |
STRE CH(89) |
|
78 |
3097 |
2976 |
18 |
15 |
63 |
18 |
STRE CH(91) |
|
77 |
3094 |
2974 |
33 |
26 |
145 |
41 |
STRE CH(87) |
|
76 |
3083 |
2963 |
8 |
6 |
24 |
7 |
STRE CH(98) |
|
75 |
3047 |
2928 |
18 |
15 |
94 |
27 |
STRE CH(97) |
|
74 |
3029 |
2911 |
19 |
15 |
114 |
32 |
STRE CH(98) |
|
73 |
2915 |
2801 |
123 |
100 |
194 |
55 |
STRE CH(96) |
|
72 |
2902 |
2789 |
31 |
25 |
50 |
14 |
STRE CH(97) |
|
71 |
2886 |
2773 |
53 |
43 |
108 |
31 |
STRE CH(96) |
|
70 |
2218 |
2131 |
4 |
3 |
282 |
80 |
STRE CC(96) |
|
69 |
1643 |
1579 |
9 |
7 |
42 |
12 |
STRE CC(38) |
|
68 |
1621 |
1558 |
1 |
1 |
10 |
3 |
STRE CC(50)+BEND CCC(12) |
|
67 |
1526 |
1466 |
12 |
10 |
1 |
0 |
BEND HCC(69)+BEND CCC(10) |
|
66 |
1518 |
1459 |
8 |
7 |
4 |
1 |
BEND HCH(68)+TORS HCCC(13) |
|
65 |
1508 |
1449 |
14 |
11 |
12 |
4 |
BEND HCH(69)+TORS HCNC(10) |
|
64 |
1502 |
1444 |
6 |
5 |
4 |
1 |
BEND HCH(66) |
|
63 |
1501 |
1443 |
5 |
4 |
8 |
2 |
BEND HCH(39) |
|
62 |
1487 |
1429 |
3 |
2 |
9 |
3 |
BEND HCH(55) |
|
61 |
1480 |
1423 |
4 |
3 |
4 |
1 |
BEND HCH(33) |
|
60 |
1480 |
1422 |
2 |
2 |
8 |
2 |
BEND HCC(10)+BEND HCH(56) |
|
59 |
1461 |
1404 |
4 |
3 |
7 |
2 |
BEND HCH(89) |
|
58 |
1412 |
1357 |
19 |
15 |
2 |
0 |
BEND HCH(83) |
|
57 |
1395 |
1341 |
9 |
8 |
6 |
2 |
BEND HCC(24) |
|
56 |
1377 |
1323 |
11 |
9 |
5 |
2 |
BEND HCC(43) |
|
55 |
1368 |
1315 |
25 |
20 |
24 |
7 |
TORS HCCC(17)+TORS HCNC(23) |
|
54 |
1353 |
1300 |
8 |
7 |
12 |
3 |
BEND HCC(35)+TORS HCNC(21) |
|
53 |
1337 |
1285 |
2 |
1 |
1 |
0 |
STRE CC(69) |
|
52 |
1312 |
1261 |
1 |
1 |
12 |
3 |
BEND HCC(24)+TORS HCCC(36)+TORS HCNC(11) |
|
51 |
1294 |
1244 |
8 |
7 |
5 |
1 |
BEND HCC(31)+TORS HCNC(22) |
|
50 |
1250 |
1201 |
42 |
34 |
1 |
0 |
STRE NC(13)+BEND HCC(10)+TORS HCNC(15) |
|
49 |
1238 |
1190 |
5 |
4 |
3 |
1 |
BEND HCC(12)+TORS HCCC(12)+TORS HCCC(11) |
|
48 |
1218 |
1170 |
11 |
9 |
35 |
10 |
STRE CC(28)+TORS HCCC(12) |
|
47 |
1203 |
1156 |
0 |
0 |
4 |
1 |
STRE CC(10)+BEND HCC(73) |
|
46 |
1181 |
1135 |
0 |
0 |
3 |
1 |
BEND HCC(75) |
|
45 |
1173 |
1127 |
16 |
13 |
13 |
4 |
STRE NC(22)+BEND HCC(19) |
|
44 |
1144 |
1099 |
14 |
11 |
2 |
1 |
BEND HCH(16)+TORS HCNC(56) |
|
43 |
1123 |
1079 |
24 |
19 |
1 |
0 |
BEND HCC(10)+TORS HCCC(10) |
|
42 |
1100 |
1057 |
12 |
10 |
2 |
1 |
TORS HCCC(11) |
|
41 |
1083 |
1041 |
28 |
23 |
3 |
1 |
STRE NC(51)+STRECC(10) |
|
40 |
1060 |
1019 |
2 |
1 |
1 |
0 |
BEND HCC(15)+ TORS HCCC(18) |
|
39 |
1050 |
1009 |
5 |
4 |
17 |
5 |
STRE CC(41) |
|
38 |
1017 |
977 |
0 |
0 |
46 |
13 |
STRE CC(22)+BEND CCC(61) |
|
37 |
1013 |
974 |
26 |
21 |
5 |
2 |
STRE CC(13)+STRE NC(15)+TORS HCCC(12)+TORS CCCN(14) |
|
36 |
1000 |
961 |
0 |
0 |
0 |
0 |
TORS HCCC(66)+TORS CCCC(14) |
|
35 |
984 |
945 |
0 |
0 |
0 |
0 |
TORS HCCC(89) |
|
34 |
976 |
938 |
18 |
14 |
4 |
1 |
STRE CC(12)+TORS HCCC(46)+TORS CCCN(18) |
|
33 |
951 |
914 |
7 |
6 |
11 |
3 |
STRE CC(34)+STRE NC(13) |
|
32 |
928 |
892 |
2 |
1 |
3 |
1 |
TORS HCCC(68) |
|
31 |
902 |
867 |
5 |
4 |
3 |
1 |
STRE CC(17)+ TORSHCCC(15) |
|
30 |
867 |
833 |
5 |
4 |
2 |
1 |
STRE CC(29)+TORS HCCC(14) |
|
29 |
856 |
823 |
0 |
0 |
0 |
0 |
TORS HCCC(98) |
|
28 |
843 |
810 |
3 |
2 |
11 |
3 |
STRE CC(10)+BEND CCC(15) |
|
27 |
808 |
777 |
11 |
9 |
4 |
1 |
STRE NC(29)+BEND CCC(14) |
|
26 |
755 |
725 |
31 |
25 |
4 |
1 |
TORS HCCC(44)+TORS CCCC(15) |
|
25 |
714 |
686 |
41 |
33 |
0 |
0 |
TORS HCCC(26)+ TORS CCCC(36)+OUT CCCC(12) |
|
24 |
701 |
674 |
44 |
35 |
9 |
2 |
BEND HCC(87)+TORS HCCC(10) |
|
23 |
664 |
638 |
53 |
43 |
7 |
2 |
BEND HCCC(10)+TORS HCCC(88) |
|
22 |
636 |
612 |
0 |
0 |
4 |
1 |
BEND CCC(16)+BEND CCC(54) |
|
21 |
628 |
604 |
5 |
4 |
2 |
1 |
BEND CCN(11) |
|
20 |
543 |
522 |
2 |
1 |
1 |
0 |
BEND CCC(12)+BEND CCN(15) |
|
19 |
518 |
498 |
11 |
9 |
1 |
0 |
OUT CCCC(24) |
|
18 |
480 |
461 |
1 |
1 |
5 |
1 |
STRE CC(12)+BEND CNC(21)+BEND CCN(16) |
|
17 |
428 |
411 |
2 |
1 |
2 |
0 |
BEND CCN(25) |
|
16 |
415 |
399 |
0 |
0 |
0 |
0 |
TORS HCCC(12)+TORS CCCC(68) |
|
15 |
411 |
395 |
3 |
2 |
2 |
1 |
BEND CCC(15)+BEND CNC(48) |
|
14 |
372 |
357 |
7 |
6 |
4 |
1 |
TORS CCCN(18)+OUT CCCN(20) |
|
13 |
362 |
348 |
1 |
1 |
3 |
1 |
BEND CCC(20)+TORS HCCC(21) |
|
12 |
321 |
308 |
2 |
2 |
3 |
1 |
TORS HCCC(40)+TORS CCCN(15) |
|
11 |
309 |
297 |
2 |
1 |
3 |
1 |
BEND CCC(20)+BEND CNC(11)+ TORS HCCC(10)+OUT CCCN(16)+OUT CCNC(10) |
|
10 |
281 |
270 |
0 |
0 |
1 |
0 |
BEND CNC(12)+BEND CCN(22)+TORS HCCC(20) |
|
9 |
247 |
238 |
0 |
0 |
2 |
0 |
STRE CC(12)+BEND CCC(10)+BEND NCC(28) |
|
8 |
237 |
228 |
2 |
1 |
2 |
0 |
BEND CCC(14)+TORS CCCC(32) |
|
7 |
205 |
197 |
1 |
1 |
1 |
0 |
TORS HCNC(70) |
|
6 |
142 |
136 |
1 |
0 |
6 |
2 |
BEND CCC(35)+BEND CCN(29) |
|
5 |
92 |
89 |
0 |
0 |
2 |
1 |
BEND CCC(28)+OUT CCCC(20) |
|
4 |
84 |
81 |
0 |
0 |
2 |
1 |
TORS CCNC(61) |
|
3 |
50 |
48 |
0 |
0 |
2 |
1 |
TORS NCCC(12)+TORS CCCC(44)+OUT CCCC(12) |
|
2 |
30 |
29 |
0 |
0 |
0 |
0 |
TORS CCNC(23)+TORS CNCC(20)+TORS NCCC(22)+OUT CCCN(12) |
|
1 |
26 |
25 |
0 |
0 |
2 |
1 |
TORS CNCC(14)+TORS NCCC(50)+TORS CCCC(15) |
Table 2: Theoretical vibrational spectroscopic data with vibrational assignments for title compound using DFT B3LYP/6-311++G (d, p) basis set.
Frontier Molecular Orbital
Frontier molecular analysis was carried out for the title compound using DFT method along with B3LYP/6-311++G (d, p) to understand the interactions of the molecule with other molecules. Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) is derived from FMO analysis. Transition between HOMO whose energy is less as they are at ground state to LUMO whose energy levels are high as they constitute excited states gives us the values of Energy Band Gap (eV) shown in fig. 4 [22,23]. Whilst Homo are electron donors, the LUMO is more electronegative in nature. High electron affinities values denote that the molecular interactions are strong. Electronegativity deals with the tendency of the molecule to accept more electrons. The energy gap between two states plays a vital role as many other parameters such as electron affinity, ionization potential, chemical softness and are tabulated in table 4. Chemical softness and chemical hardness were found to be 0.17689 η and 2.8265 S. HOMO and LUMO values characterizes the chemical kinetics stability of the title compound. For shorter energy gaps, the compound is polarized, and they are called as soft molecules [24].
| Basis Set | B3LYP/6-311++G (d, p) |
| ELUMO(eV) | -0.4493 |
| EHUMO(eV) | -6.1022 |
| Ionization potential(I) | 6.1022 |
| Electron affinity(A) | 0.4493 |
| Energy gap(eV) | 5.6529 |
| Electronegativity(χ) | 3.2757 |
| Chemical potential(μ) | -3.2757` |
| Chemical hardness(η) | 2.8265 |
| Chemical softness(S) | 0.1769 |
Table 4: Calculated energies values of the title compound.
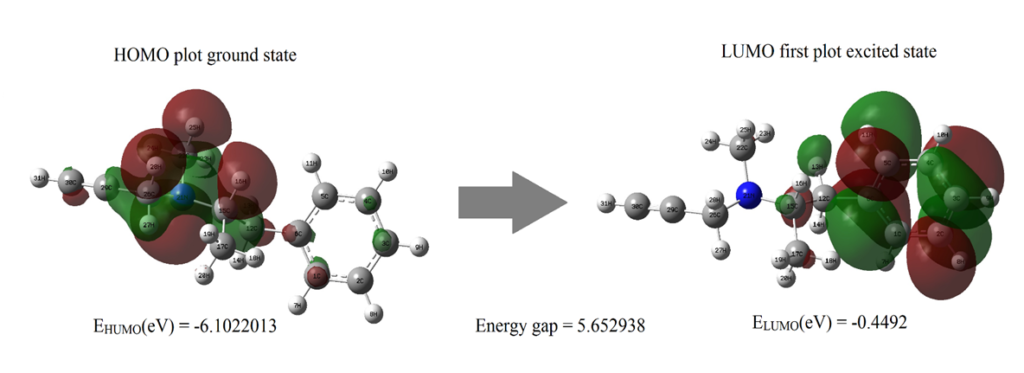
Figure 4: The molecular orbitals and energies for HOMO and LUMO of ethyl (2R)-N-methyl-1-phenyl-N-prop-2-ynylpropan-2-amine
UV – Visible Spectral Analysis
UV spectrum of the title compound was obtained from GAUSSUM software. Time dependent DFT was carried out to visualise the effect of solvent to aqueous phase of the title compound. The study was carried out in solvent phase in solvent model density (SDM). Through the computational method, the values of maximum wavelength λmax (nm), Band gap energy(eV), excitation energy (cm-1), and oscillator strength (f) of the title compound were obtained and tabulated in table 5 [25]. The band gap was calculated using the formula E = hc/λ. UV spectral analysis show the electronic absorption which excites the atom from highest occupied molecular orbital (HOMO) to lowest unoccupied molecular orbital (LUMO). This energy gap (5.0371eV) between the two molecular orbitals agreed with the energy band gap (5.6529eV) calculated from FMO [26]. Fig .5 Shows theoretical UV–vis spectrum of the title compound.
| λmax (nm) | Band gap (eV) | Energy (cm-1 ) | f | Assignments |
| 246.1696 | 5.037177 | 40622.39 | 0.0341 | HOMO->LUMO (90%) |
| 240.9549 | 5.146192 | 41501.54 | 0.0006 | HOMO->L+1 (93%) |
| 233.6135 | 5.30007913 | 42805.75 | 0.0036 | H-2->LUMO (20%), H-2->L+1 (17%), H-1->LUMO (29%), H-1->L+1 (26%) |
Table 5: Theoretically calculated electronic properties using the TD-DFT method.
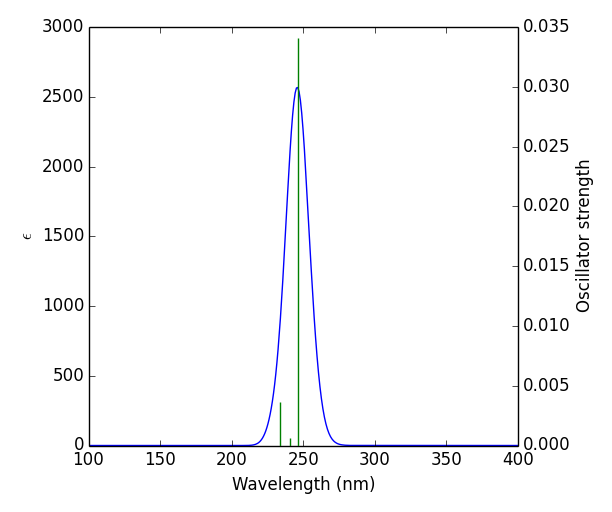
Figure 5: theoretical UV–vis spectrum of the title compound
Molecular Electrostatic Potential
The distribution of charges over a 3-dimensional space creates an electrostatic potential around it. MEP also known as molecular electrostatic potential explains us about how the molecule would react to charges surrounding it. The mapping of MEP was done with DFT methods along with basis set B3LYP/6-311++G (d, p) [27]. There are two major sites in a molecule namely electrophilic sites wherein the atoms in this region have tendency to attract electrons and nucleophilic sites wherein the atoms in this region are ready to share electrons. MEP diagram is shown in Fig.6. The colour region varies from red to blue where red signifies electrophilic site and blue signifies nucleophilic site. Potential in the current study ranges from – 4.002e-2 eV to 4.002e-2 eV. From the figure, the red region denotes electrophilic region which is due to the presence of N21 atom [28,29]. The blue region at the borders constitutes nucleophilic sites. H atoms occupying this region are nucleophilic in nature. The green site in map indicates neutral sites.
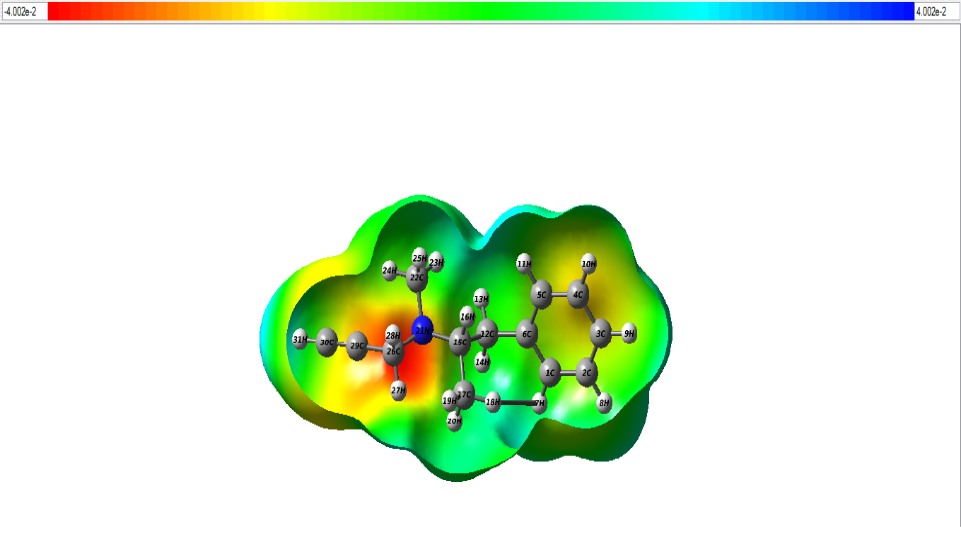
Figure 6: The MEP map of the title compund.
Electron Localization Function & Local Orbital Locator
Topological analyses of ELF and LOL were carried out using MULTIWFN software [30]. The 2D mappedELF and LOL is shown in fig. 7 and fig. 8. The colour codes vary from 0.001 to 1.000. High ELF values are represented by red region whilst low ELF values are represented by blue region [31]. The main objective to perform ELF studies is to understand quantitative behaviour of electrons in a system. The difference in kinetic energy density contributes towards Pauli repulsion on two like spin electrons which attributes towards the behaviour of the electron. The red region indicates high Pauli repulsion, and blue region indicates low Pauli repulsion [32]. Several colours are represented on this surface. For the title compound, blue colour circle in 2d structure, indicates the presence of a depletion region between valence shell and inner shell.
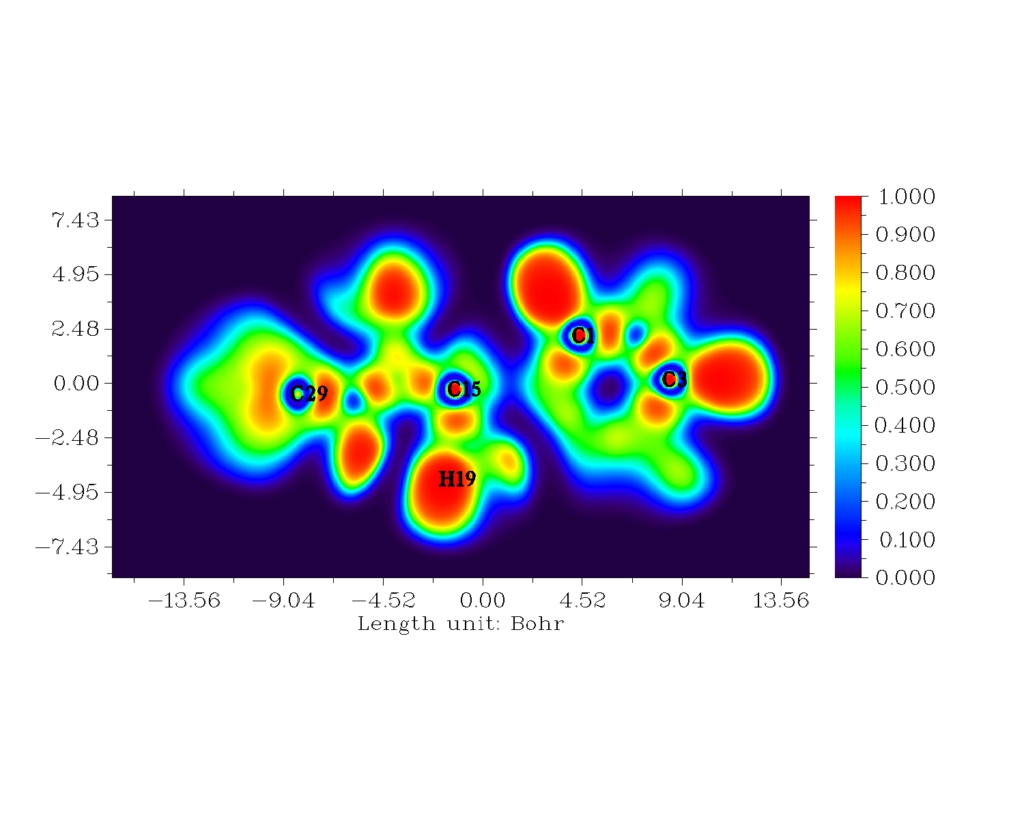
Figure 7: The 2-D mapped ELF for the title compound.
The red orange region depicts strong electronic localisation [33,34]. Localized orbital locator is like that of ELF. The hydrogen and carbon regions have minimum values of LOL. The LOL has colour codes ranging from 0.000 to 0.800 [35]. The white region encircled with red region of H19 atom indicates that there is an excess of electron cloud which exceeds the covalent region.
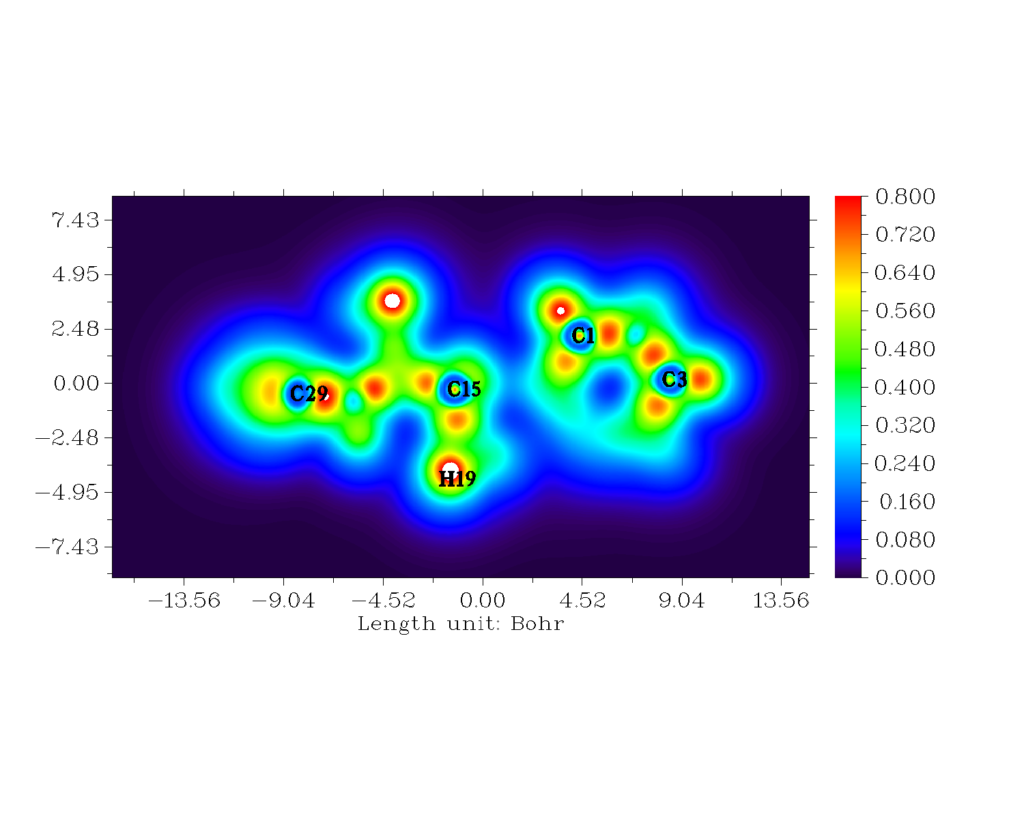
Figure 8: The 2-D mapped LOL for the title compound.
Drug Likeness
The canonical smile of selegiline was taken from PubChem [36]. Using this, Drug likeness parameters tabulated in table 8 was obtained by SwissADME software [37]. The acceptable values of HBA and HBD should be less than 5 and 10 respectively, which is 1 and 0 for the title compound. The MlogP parameter gives an idea about lipophilic character of the molecule, which is 3.25 for the title compound. This is in the acceptable range as it is less than 4.15. The number of rotatable bonds should be less than 10 and is 4 in this case. Based on the analysis, the title compound satisfies Lipinski’s rule of five most likely favourable to be subjected to any desired studies since it proves that it is an active drug. [38]
| Descriptors | Value |
| Hydrogen bond donors (HBD) | 0 |
| Hydrogen bond acceptors (HBA) | 1 |
| ALogP | 3.25 |
| Polar surface area (PSA) Å2 | 3.24 |
| Molar refractivity | 61.31 |
| Number of atoms | 31 |
| Number of rotatable bonds | 4 |
Table 8: Descriptors and corresponding values for drug likeness of the title compound.
Molecular Docking
Molecular docking of the title compound was carried out using AutoDock software. Protein used for docking was taken from protein data bank (PDB). Optimised docked structure of target protein with the title compound is shown in fig. 9. Protein had a resolution of 2.30 Å [39]. Protein structure was obtained from RCSB PDB format. Auto dock tool (ADT), a graphical user interface was used to obtain binding energy of the title compound to a receptor. The title compound was selected to be docked into the active site of protein 4f1t (Parkinson’s disease). Docking parameters such as Bond distance (Å), Inhibition constant (μm), Intermolecular energy (kcal/mol), Binding Energy (kcal\mol) and Reference RMSD (Å) are tabulated in table 7. Minimum binding energy of – 3.61 kcal/mol and intermolecular energy of −4.8 kcal/mol have been observed in the interaction which interprets that protein and the title compound has strong bond between them. Inhibition constant and bond distance found to be 2.27 μm and 2.631 Ǻ respectively [40].
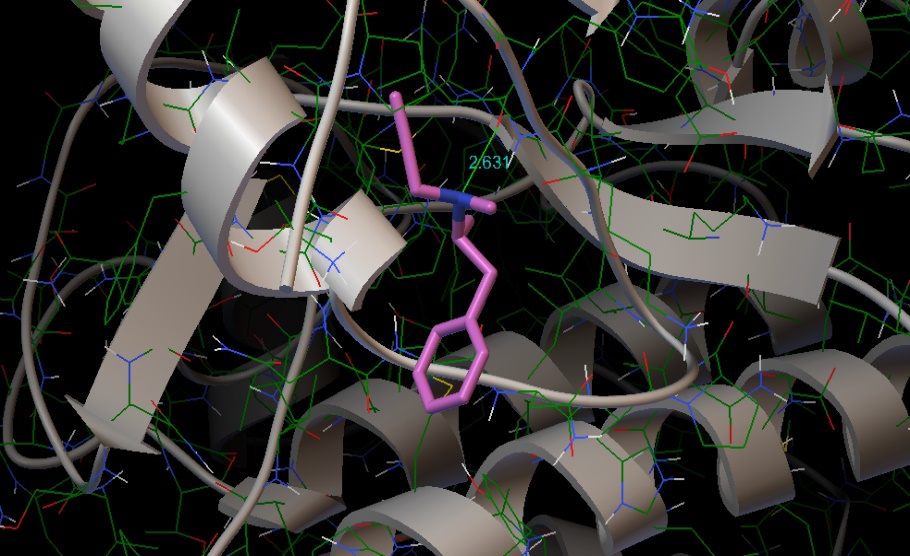
Figure 9: Optimised docked structure of the target protein with the title compound.
| Protein | Bonded residues | Bond distance Å | Inhibition constant μm | Intermolecular energy kcal/mol | Binding Energy kcal\mol | Reference RMSD Å |
| 4f1t | LEU1093 | 2.631 | 2.27 | -4.8 | -3.61 | 19.67 |
Table 7: Docking parameters of the protein 4f1t with the title compound.
Conclusion
In the present work, a detailed Spectroscopic (FT-IR, FT-RAMAN), UV, FMO, Reactive site analysis (MEP, ELF, LOL) along with molecular docking studies on the title compound, Selegiline has been reported. Optimized molecular geometry was obtained using DFT method along with basis set B3LYP/6-311++G (d, p). The complete vibrational assignments and calculations of potential energy distribution (PED) were carried out using Veda software. Reactive sites of the title compound were determined using LOL and ELF. From MEP diagram, the electrophilic and nucleophilic sites were identified. Electronic properties of the compound were studied theoretically using TD-DFT method. Energy gap calculated from UV spectrum was in good agreement with HOMO-LUMO energy gap. Finally, molecular docking analysis was carried out for protein 4flt associated with Parkinson’s disease and showed binding affinity value of -3.61 kcal/mol.
References
- Youdim MDH, Riederer PF (2004) A review of the mechanisms and role of monoamine oxidase inhibitors in Parkinson’s disease. Neurology 63: S32-S35.
- Youdim MDH, Edmondson D, Tipton KF (2006) The therapeutic potential of monoamine oxidase inhibitors. Nature reviews neuroscience 7: 295-309.
- Gerlach M, Youdim MBH, Riederer P (1996) Pharmacology of selegiline. Neurology 47: 137S-145S.
- Ward CD (1994) Does selegiline delay progression of Parkinson’s disease? A critical re-evaluation of the DATATOP study. Journal of Neurology, Neurosurgery & Psychiatry 57: 217-220.
- Salonen JS, Nyman L, Boobis AR, Edwards RJ, Watts P, et al. (2003) Comparative studies on the cytochrome p450-associated metabolism and interaction potential of selegiline between human liver-derived in vitro systems.” Drug metabolism and disposition 31: 1093-1102.
- Gnanasambandan T, Gunasekaran S, Seshadri S (2013) Quantum Chemical and Spectroscopic (FT-IR, FT-Raman) Study, First Order Hyperpolarizability, NBO, Analysis HOMO and LUMO Analysis of Selegiline by abinitio HF and DFT Method. Oriental Journal of Chemistry 29: 185.
- Becke AD (1994) Density-fumctional exchange energy approximation with correct asymptotic behavior. Phys. Rev. B: 54.
- Perdew JP, Burke K, Wang Y (1996) Generalized gradient approximation for the exchange-correlation hole of a many electron system. Phys. Rev. B: 54.
- Chemcraft – graphical software for visualization of quantum chemistry computations.
- Jamroz MH (2004) Vibrational Energy Distribution Analysis: VEDA 4 Program, Warasaw, Poland.
- Roy D, Keith T, Millam J, GaussView, Semichem Inc., Shawnee Mission, KS, 2009. Version 5.
- Politzer P, Truhalr DG (Eds.), Chemical Application of Atomic and Molecular Electrostatic Potential, Plenum, Newyork, 1981.
- Mulliken RS (1934) A new electronaffinity scale; Together with Data on valence states and on valence ionization potentials and electron affinities. J. Chem. Phys. 2: 782-793.
- Tian Lu, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J. Comp. Chem 33: 580-592.
- Antoine D, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules.” Scientific reports 1: 1-13.
- Tanchuk V, Tanin V, Vovk A (2013) Multithreaded version of AutoDock 4.2 suitable for massive virtual screening of potential biologically active compounds (enzyme inhibitors).” Third International Conference” High Performance Computing” HPC-UA.
- Fathima RB, Johanan CP, Muthu S (2017) Spectroscopic investigation (FT-IR, FT-Raman, UV, NMR), Computational analysis (DFT method) and Molecular docking studies on 2-[(acetyloxy) methyl]-4-(2-amino-9h-purin-9-yl) butyl acetate. Int. J. Mater. Sci 12: 196-210.
- WebSpectra.
- IR Spectroscopy Tutorial: Amines.
- Barnes AJ, Majid MA, Stuckey MA, Gregory P, Stead CV (1985) The resonance Raman spectra of Orange II and Para Red: molecular structure and vibrational assignment. Spectrochimica Acta Part A: Molecular Spectroscopy 41: 629-635.
- Joe IH, Kostova I, Ravikumar C, Amalanathan M, Cîntǎ Pînzaru S (2009) Theoretical and vibrational spectral investigation of sodium salt of acenocoumarol. Journal of Raman Spectroscopy: An International Journal for Original Work in all Aspects of Raman Spectroscopy, Including Higher Order Processes, and also Brillouin and Rayleigh Scattering 40: 1033-1038.
- Ramazani A, Sheikhi M, Yahyaei H (2017) Molecular Structure, NMR, FMO, MEP and NBO Analysis of Ethyl-(Z)-3-phenyl-2-(5-phenyl-2H-1, 2, 3, 4-tetraazol-2-yl)-2-propenoate Based on HF and DFT Calculations. Chemical Methodologies 1: 28-48.
- Rizwana F, Prasana JC, Muthu S, Abraham CS (2019) Molecular docking studies, charge transfer excitation and wave function analyses (ESP, ELF, LOL) on valacyclovir: a potential antiviral drug. Computational biology and chemistry 78: 9-17.
- Manjusha P, Prasana JC, Muthu S, Rizwana FB (2019) A computational and spectroscopic interpretation (FT-IR, FT-Raman, UV–vis and NMR) with molecular docking studies on 3-carboxy-2-hydroxy-N, N, N-trimethyl-1-propanaminium hydroxide: A pharmaceutical drug. Chemical Data Collections 20: 100191.
- Geoffrey BAS, Prasana JC, Muthu S, Abraham CS, et al. (2019) Spectroscopic and quantum/classical mechanics based computational studies to compare the ability of andrographolide and its derivative to inhibit nitric oxide synthase. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 218: 374-387.
- Fathima Rizwana B, Johanan CP, Muthu S (2017) Spectroscopic investigation (FT-IR, FT-Raman, UV, NMR), Computational analysis (DFT method) and Molecular docking studies on 2-[(acetyloxy) methyl]-4-(2-amino-9h-purin-9-yl) butyl acetate. Int. J. Mater. Sci 12: 196-210.
- Marinho ES, Marinho MM (2016) A DFT study of synthetic drug topiroxostat: MEP, HOMO, LUMO.” International Journal of Scientific & Engineering Research 7: 8.
- Fathima Rizwana B, Muthu S, Johanan CP, Abraham CS, Raja M (2018) Spectroscopic (FT-IR, FT-Raman) investigation, topology (ESP, ELF, LOL) analyses, charge transfer excitation and molecular docking (dengue, HCV) studies on ribavirin. Chemical Data Collections 17: 236-250.
- Prabakaran M, Prasana JC (2021) Molecular docking studies, natural bond orbital, charge transfer excitation, NLO, ELF, LOL, and drug likeness analysis on Cefixime: a potential anti-bacterial effective. Sci Academique 2: 1-18.
- George J, Prasana JC, Muthu S, Kuruvilla TK, Saji RS, et al. (2020) Evaluation of vibrational, electronic, reactivity and bioactivity of propafenone—A spectroscopic, DFT and molecular docking approach. Chemical Data Collections 26: 100360.
- Noureddine O, Issaoui N, Medimagh M, Al-Dossary O, et al. (2021) Quantum chemical studies on molecular structure, AIM, ELF, RDG and antiviral activities of hybrid hydroxychloroquine in the treatment of COVID-19: Molecular docking and DFT calculations. Journal of King Saud University-Science 33: 101334.
- Saji RS, Prasana JC, Muthu S, George J, Kuruvilla TK, et al. (2020) Spectroscopic and quantum computational study on naproxen sodium. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 226: 117614.
- Radder SB, Melavanki R, Hiremath SM, Kusanur R, et al. (2021) Synthesis, spectroscopic (FT-IR, FT-Raman, NMR & UV-Vis), reactive (ELF, LOL, Fukui), drug likeness and molecular docking insights on novel 4-[3-(3-methoxy-phenyl)-3-oxo-propenyl]-benzonitrile by experimental and computational methods. Heliyon 7: e08429.
- Pilepić V, Uršić S (2001) Nucleophilic reactivity of the nitroso group. Fukui function DFT calculations for nitrosobenzene and 2-methyl-2-nitrosopropane. Journal of Molecular Structure: THEOCHEM 538.1-3: 41-49.
- Manju P, Muthu S, Gowda NMN (2017) Quantum mechanical and spectroscopic (FT-IR, FT-Raman, 1H, 13C NMR, UV-Vis) studies, NBO, NLO, HOMO, LUMO and Fukui function analysis of 5-Methoxy-1H-benzo [d] imidazole-2 (3H)-thione by DFT studies. Journal of Molecular Structure 1130: 511-521.
- Compound Summary. Selegiline
- http://www.swissadme.ch/index.php
- Saji RS, Prasana JC, Muthu S, George J, Kuruvilla TK, et al. (2020) Spectroscopic and quantum computational study on naproxen sodium. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 226: 117614.
- https://www.rcsb.org/structure/4F1T
- Manjusha P, Prasana JC, Muthu S, Rizwana BF (2019) A computational and spectroscopic interpretation (FT-IR, FT-Raman, UV–vis and NMR) with molecular docking studies on 3-carboxy-2-hydroxy-N, N, N-trimethyl-1-propanaminium hydroxide: A pharmaceutical drug. Chemical Data Collections 20: 100191.