

Molecular docking studies, natural bond orbital, charge transfer excitation, NLO, ELF, LOL, and drug likeness analysis on Cefixime: a potential anti-bacterial effective Manjusha Prabakaran1,2, Johanan Christian Prasana2*
1Department of Physics, S.D.N.B Vaishnav College for Women, Chromepet and University of Madras, Chepauk, Chennai, Tamilnadu, India
2Department of Physics, Madras Christian College, Chennai, Tamilnadu, India
*Correspondence to: Johanan Christian Prasana
Citation: Prabakaran M and Prasana JC (2021) Molecular docking studies, natural bond orbital, charge transfer excitation, NLO, ELF, LOL, and drug likeness analysis on Cefixime: a potential anti-bacterial effective. Sci Academique 2(1): 50-67.
Received: 04 May, 2021; Accepted: 24 May 2021; Publication: 28 May 2021
Abstract
In this present study, a pharmaceutically potent property of the title compound has been analysed by theoretical analysis with higher order basis set B3LYP/6-311++ G (d, p). Chemical reactivity of the titled compound was confirmed using ELF and LOL studies. Theoretical simulations through two different module CAM-B3LYP/6-31G (d, p) and B3LYP/6-311++G(d,p) gas phase have been carried out for better comparison. NBO studies helpful understanding the stability and charge delocalization of the titled compound. All the results and studies were presented best, after thorough literature survey. Molecular docking studies have been carried out to compliment the pharmaceutically potent nature of the title compound.
Keywords: DFT; Molecular docking; ELF; LOL; NLO; NBO
Introduction
(6R,7R)-7-{[2-(2-Amino-1,3-thiazol-4-yl)-2-(carboxymethoxyimino)acetyl]amino}-3-ethenyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (6CA2C), a cephalosporin antibiotic is used to treat various bacterial infections such as strep throat, pneumonia, urinary tract infections and gonorrhoea [1-4]. The bactericidal action of 6CA2C is due to inhibition of cell wall synthesis. It binds to penicillin binding proteins (PBP) leading to final transpeptidation step of the peptidoglycan synthesis. During this process, the bacterial cell activity is constrained due to inhibition of biosynthesis, arresting cell wall assembly [5-7]. Some significant microorganisms such as Escherichia coli and Proteus mirabilis cause urinary tract infections well treated by 6CA2C. After administering the title compound, inflammatory ear infection Otitis media, Tonsillitis and pharyngitis throat infection caused by Haemophilus influenzae, Moraxella catarrhalis and Streptococcus pyogenes are treated [8-11].
A thorough literature survey has been performed on 6CA2C based on bacterial infections and it is found that no work has been carried out on spectroscopic analysis and DFT [1]. Therefore, theoretical DFT calculations featuring nonlinear optical property, NBO, wavefunction analyses, charge transfer due to excitation, molecular docking and drug likeness have been carried out for the title compound.
Materials and Methods
The titled compound 6CA2C is purchased from Sigma-Aldrich chemical company (USA) with 99% pureness and used the same form to record UV-Visible spectra. Ultraviolet absorption spectrum is recorded for DMSO as solvent using JASCO V-670 Spectrometer in the range 200-800 nm sophisticated instrumentation facility (SIF), Vellore Institute of Technology, Vellore, India. Computational calculations were set up based on Gaussian 09 version [12] with the higher order basis set B3LYP/6-311++G (d,p) for vibrational assignments. Density functional theory (DFT) [13-17] also called as computational modelling method is used to investigate the electronic structure of many body systems Origin version 6.1 is used to plot the comparative graphs for spectroscopic characterizations. Theoretical UV spectra have been created with two different modules for better calculations and plotted. Using NBO output, density of states (DOS), partial density of states (PDOS) and crystal overlap population (COOP) were imaged using Gauss Sum software [18]. Multiwfn 5.0 [19] is used to generate electron localized function (ELF) and local orbital locator (LOL) maps for topological studies. Autodock tools 1.5.6 (ADT) software is used to model the ligand and protein mode of interactions for three different active site proteins captured in 3D images.
Results and Discussions
Electronic properties
Density of States (DOS, PDOS and COOP) analysis
Density of states (DOS) spectrum demonstrates the number of available states as a function of energy. Figure 1 represents the optimized molecular structure used for all the calculations. Figure 2 (a) corresponds to DOS spectrum in which maximum states under virtual orbitals (negative) and occupied orbitals (positive). Partial DOS (PDOS) spectrum corresponds to the energy range -20 to 10 eV with fragments C-H and C-S maximum. C-C and O-H are considered to be the least among the other contributions and the Figure 2 (b) indicates the spectrum of partial density of states. Crystal orbital overlap population (COOP) indicates the nature of overlap of between two groups of atoms. Furthermore, Figure 2 (c) stipulates the overlap of C-N with C-S as maximum anti-bonding interactions represented in negative y axis. This study reveals about composition of molecular orbitals and chemical bonding of the title compound.
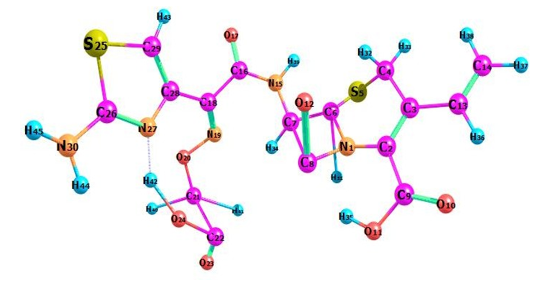
Figure 1: Optimized geometrical figure of 6CA2C
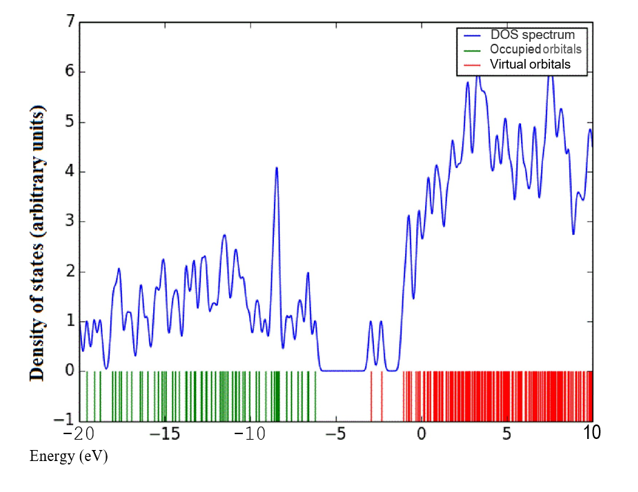
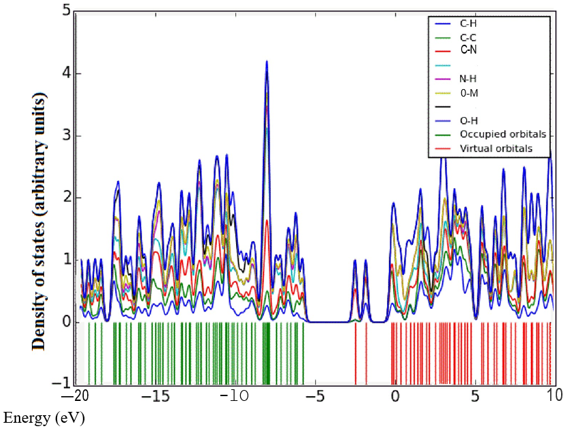
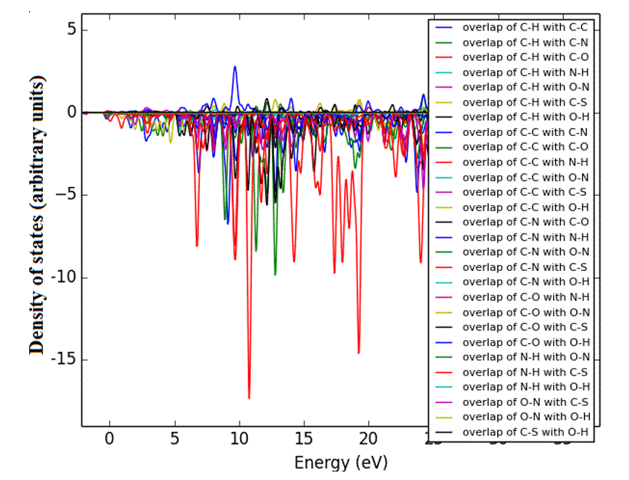
Figure 2(a-c): DOS, PDOS and COOP spectrum of 6CA2C.
Charge transfer due to excitation
Electron-hole distribution for the three excited states S1, S2 and S3 are represented in Figure 3. Details pertaining to charge transfer length D, o v e r l a p i n t e g r a l S, excitation energy ΔE as well as Δr of excited states S1, S2 and S3 are tabulated in Table 1. A small overlap integral S, large charge transfer length D and ∆r index greater than 2.0 Ǻ affirms there is a stronger charge transfer at the third excited state. Also, RMSD electron distribution relates to broader orientation in X direction compared to Y and Z directions for all three states.t index for states S2 and S3 are not negative in all the directions projecting a medium spatial separation whereas state S1 is negative values in all the directions showing hole-electron overlap.
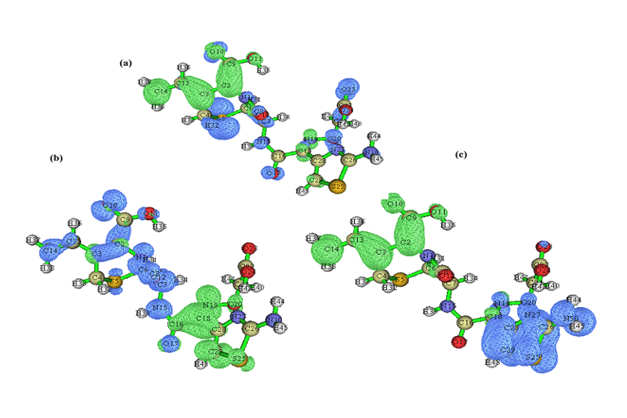
Figure 3: Electron-hole distribution for the excited states a) S1 b) S2 and c) S3 of 6CA2C.
|
Parameters |
Excited states |
||
|
S1 |
S2 |
S3 |
|
|
Overlap integral of electron-hole S |
0.187 |
0.228 |
0.114 |
|
Charge transfer length D(Ǻ) |
2.902 |
3.625 |
7.614 |
|
Excitation energy, ∆E (eV) |
3.095 |
3.529 |
3.701 |
|
∆r (Ǻ) |
8.954 |
9.251 |
14.332 |
|
RMSD of electron |
|
|
|
|
X |
3.007 |
3.228 |
3.025 |
|
Y |
1.482 |
1.419 |
1.482 |
|
Z |
1.186 |
1.394 |
1.189 |
|
RMSD of hole |
|
|
|
|
X |
3.161 |
2.748 |
1.538 |
|
Y |
2.087 |
1.504 |
1.388 |
|
Z |
1.384 |
1.386 |
1.45 |
|
t index |
|
|
|
|
X |
-0.571 |
0.483 |
5.300 |
|
Y |
-1.530 |
-0.798 |
-0.814 |
|
Z |
-0.145 |
-0.584 |
-0.975 |
Table 1: Overlap integral, charge transfer length, ∆r and excitation energy for different excited states of 6CA2C.
UV-visible spectral analysis
Theoretical and experimental spectra of ultraviolet-visible analysis are represented in Figure 4 and wavelength, bandgap and oscillator strength are listed in Table 2. Density functional calculations are used to identify the excitation region of the compound through ultraviolet-visible analysis [20-22]. Both experimental DMSO and density functional theory method using CAM-B3LYP/6-31 G (d,p) and Gas phase modules are in concordance. Highest occupied molecular orbital to lowest unoccupied molecular orbital transition for 6CA2C is 3.867 electron volts [23,24] and energy gap obtained through experimental solvent DMSO is 3.737 electron volts, respectively. Time dependent- density functional theory gives the bandgap as 3.737 electron volts for solvent DMSO by CAM-B3LYP/6-31G (d,p) module. The peak wavelengths are observed at 393, 355 and 318 nm for gas phase shows good agreement with experimental findings.
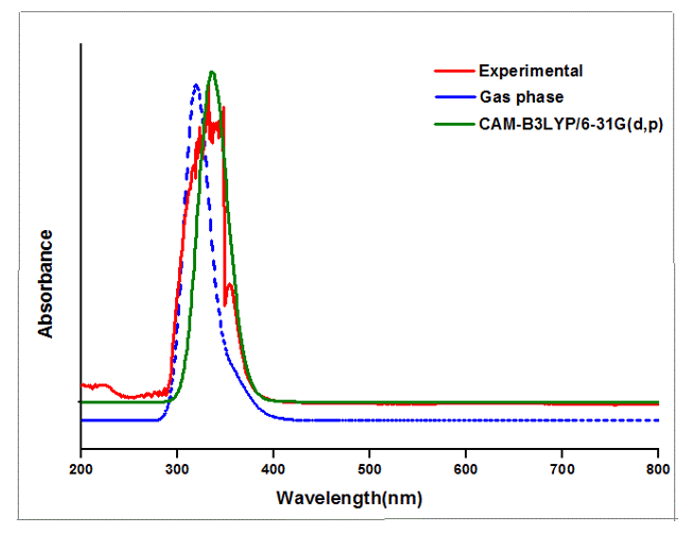
Figure 4: Experimental and theoretical UV-Vis spectrum of 6CA2C.
|
Experimental (DMSO) |
Theoretical |
||||||||||
|
CAM-B3LYP/6-31G(d,p) Solvent Phase |
B3LYP/6-311++G(d,p) Gas Phase |
||||||||||
|
λmax (nm) |
Band gap (eV) |
λcal (nm) |
Band gap (eV) |
Energy (cm-1) |
Oscillatory strength |
Assignments |
λcal (nm) |
Band gap (eV) |
Energy (cm-1) |
Oscillatory strength |
Assignments |
|
348 |
3.565 |
389 |
3.187 |
25659 |
0.0003 |
H-2->LUMO (83%) H-3->LUMO (9%) |
393 |
3.155 |
25433 |
0.0004 |
H-3->LUMO (58%), H-2->LUMO (30%), H-5->LUMO (6%) |
|
332 |
3.737 |
343 |
3.615 |
29131 |
0.0582 |
H-3->L+1 (30%), H-1->L+1 (48%), H-2->L+1 (8%), H-1->LUMO (2%), HOMO->L+1 (7%) |
355 |
3.492 |
28119 |
0.0102 |
H-3->L+1 (14%), H-2->L+1 (57%), HOMO->L+1 (24%) |
|
323 |
3.841 |
332 |
3.737 |
30065 |
0.1429 |
HOMO->LUMO (90%) H-1->LUMO (5%) |
318 |
3.899 |
31384 |
0.0795 |
H-1->LUMO (73%), HOMO->LUMO (21%) |
Table 2: Comparison of electronic properties of 6CA2C obtained theoretically and experimentally.
Topology analyses
Electron localized function (ELF)
A two dimensional image of Electron localized function is presented in Figures 5 (a) and b) starting from the scale 1.0 to 0.0. Colour codes starts from red for high ELF values, yellow to green for medium and blue shades for low ELF values respectively. Greater the electron localization in a particular region resembles in high ELF values, while minimum repulsion tends show up at low ELF values [25,26]. Carbon atoms in the ring structure namely 2 C, 6 C and 8C exhibit low localization values as represented in blue region. Sulphur atoms show a moderate ELF values and occur at neutral green region.
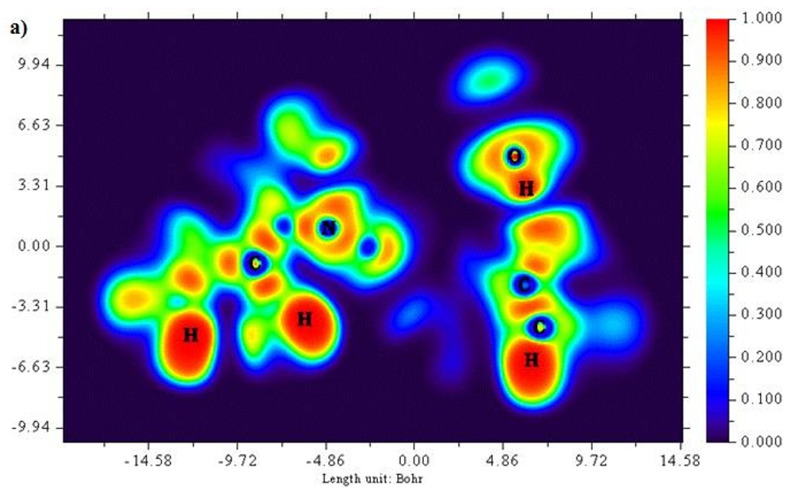
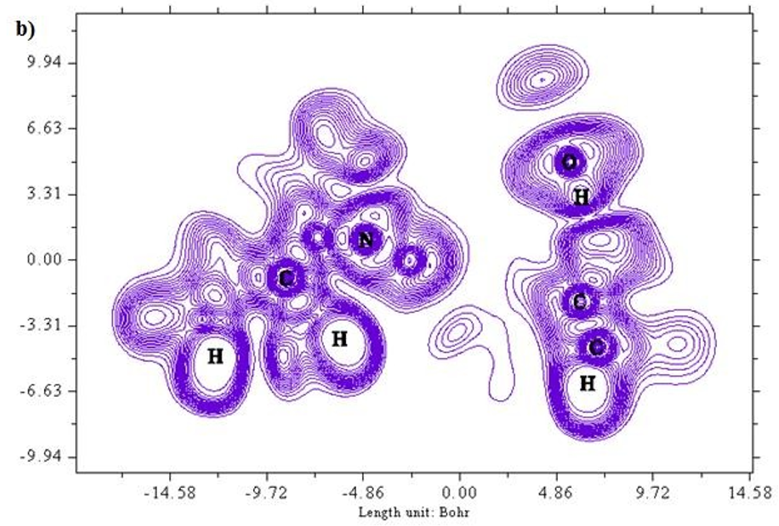
Figure 5 (a & b): ELF, color filled and contour map of the 6CA2C.
Local orbital locator (LOL)
A two-dimensional representation of localized orbital locator (colour shade and contour maps) is displayed in Figures 6 (a) and (b) with the scale range of 0.0 to 0.8. Localized orbital locator distribution is based on B3LYP/6-311++G (d,p) constitutes molecular orbitals that are limited to spatial region. Due to localized orbitals overlapping, the gradient of localized orbitals reaches its maximum as represented by red colour indication in colour shade map [27]. Most of the carbon atoms are enclosed by blue colour circles which indicate electron depletion between valence shell and inner shell. Local orbital locator shows more precise mappings compared to electron localized function. This analysis helps in understanding overlap pattern of localized orbitals of the titled compound.
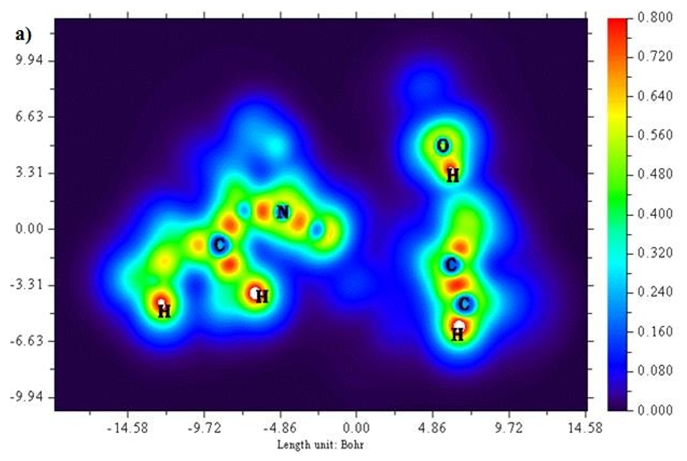
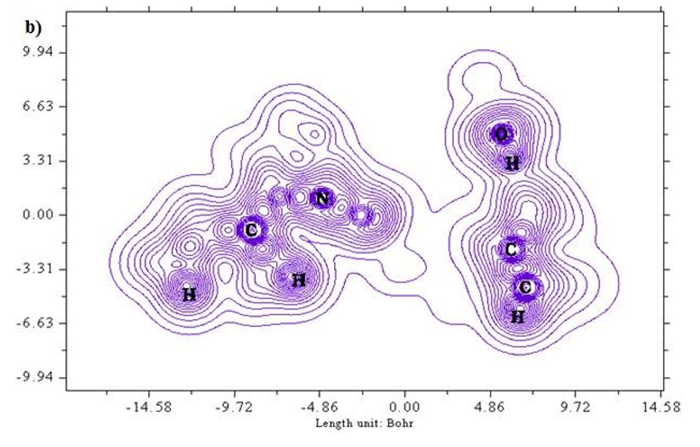
Figure 6 (a & b): LOL, colour filled and contour map of 6CA2.
Nonlinear optical properties (NLO)
A three-dimensional representation of all these parameters is listed in Table 3. Dipole moment correlates with the charge transfer is maximum at x direction compared to y and z directions. A standard nonlinear optical material Urea [28] is used for comparative study due to its intra molecular charge movement capacity. Polarizability 𝛼 (4.41×10-23 e.s.u) is 9 times greater than standard NLO material urea (0.491×10-23 e.s.u). Likewise, first order hyperpolarizability (4.36×10-30 e.s.u) is 4.65 times greater than urea (0.928×10-30 e.s.u). This confirms the title compound as a good NLO candidate.
|
Property |
Parameter |
Theoretical |
|
|
6CA2C |
Urea |
||
|
Polarizability (𝛼) |
αxx |
381.6 |
37.245 |
|
αxy |
-42.9 |
-0.194 |
|
|
αyy |
235.1 |
37.988 |
|
|
αxz |
-43.6 |
0.052 |
|
|
αyz |
-62.4 |
-0.063 |
|
|
αzz |
276.0 |
24.012 |
|
|
α (a.u) |
297.6 |
33.081 |
|
|
α (e.s.u) |
4.41×10-23 |
0.491×10-23 |
|
|
Dipole moment (𝜇) |
μx |
3.084 |
-0.806 |
|
μy |
1.384 |
1.543 |
|
|
μz |
-1.729 |
-0.008 |
|
|
μ(D) |
3.797 |
-1.741 |
|
|
First hyperpolarizability (𝛽) |
βxxx |
-59.8 |
23.748 |
|
βxxy |
-325.2 |
17.376 |
|
|
βxyy |
-11.6 |
-55.468 |
|
|
βyyy |
375.3 |
44.220 |
|
|
βzxx |
295.6 |
-0.489 |
|
|
βxyz |
243.4 |
0.034 |
|
|
βzyy |
-158.1 |
-0.531 |
|
|
βxzz |
-368.1 |
-19.037 |
|
|
βyzz |
151.5 |
33.038 |
|
|
Βzzz |
9.6 |
-1.062 |
|
|
βo (a.u) |
505.4 |
107.407 |
|
|
βo (e.s.u) |
4.36×10-30 |
0.928×10-30 |
|
Table 3: First order hyperpolarizability, static polarizability and Dipole moment values of 6CA2C.
Natural Bond orbital (NBO) analysis
Natural bond orbital analysis is a prominent study of acceptor and donor interactions with respect to stabilization energies. 𝜋 (C2-C3) corresponds to 𝜋*(C9-O10) and 𝜋*(C13-C14) has the maximum stabilization energies 25.76 and 14.79 Kcal/mol, respectively. 𝜋 (C4-S5) transition to 𝜋*(C2-C3) and 𝜋*(C3-C4) with stabilization energies 14.58 and 11.58 Kcal/mol. 𝜋 (C6-C7) has donor interactions with acceptor groups 𝜋*(C8- O12) and 𝜋*(N15-C16) at 11.04 and 12.04 Kcal/mol stabilization energies. 𝜋 (C18-N19) donor corresponds to 𝜋*(C28-C29) acceptor and 𝜋*(C16-O17) with stabilization energies 11.61 and 18.07 Kcal/mol. Lone pair donor orbitals corresponds to the interaction of O 10, N 1 to acceptors 𝜎*(C9) and (C2-C3) with 12.39 kcal/mol and 11.21 kcal/mol respectively. Table 4 depicts the stabilization energies with donor and acceptor groups of title compound. This delocalization energies at its maximum are related to intramolecular hyperconjugation allows electron in each π bonding orbital electrons to delocalize anti- bonding orbitals. This NBO analysis helps in understanding the electron density transfer from donor to acceptor and intermolecular interactions of the title compound.
|
Donor |
Type |
ED/e |
Acceptor |
Type |
ED/e |
E(2) |
E(j)-E(i) |
F(i,j) |
|
C 2 – C 3 |
π |
1.74453 |
C 9 – O 10 |
π* |
0.36369 |
25.76 |
0.32 |
0.098 |
|
C 13 – C 14 |
π* |
0.05934 |
14.79 |
0.34 |
0.067 |
|||
|
C 2 – C 9 |
σ |
1.96911 |
N 1 – C 2 |
σ* |
0.25885 |
3.97 |
1.37 |
0.066 |
|
C 4 – S 5 |
π |
1.93643 |
C 2 – C 3 |
π |
0.36511 |
14.58 |
0.66 |
0.095 |
|
C 3 – C 4 |
π* |
0.35412 |
11.58 |
1.01 |
0.036 |
|||
|
C 6 – C 7 |
π |
1.9618 |
C 8 – O 12 |
π* |
0.03181 |
11.04 |
1.23 |
0.104 |
|
N 15 – C 16 |
π* |
0.25326 |
12.04 |
1.08 |
0.03 |
|||
|
C 18 – N 19 |
π |
1.8964 |
C 28 – C 29 |
π* |
0.38277 |
11.61 |
0.47 |
0.071 |
|
C 16 – O 17 |
π* |
0.36854 |
18.07 |
0.38 |
0.08 |
|||
|
C 13 – C 14 |
σ |
1.84133 |
C 2 – C 3 |
σ* |
0.36511 |
3.12 |
0.29 |
0.083 |
|
C 18 – C 28 |
σ |
1.85649 |
C 18 – N 19 |
σ* |
0.35631 |
3.34 |
1.38 |
0.061 |
|
N 19 – O 20 |
σ |
1.85471 |
C 7 – H 34 |
σ* |
0.25897 |
0.55 |
1.59 |
0.028 |
|
C 16 – O 17 |
π |
1.96979 |
C 18 – N 19 |
π* |
0.3175 |
10.04 |
0.39 |
0.06 |
|
N 1 – C 8 |
σ |
1.97294 |
N 1 – C 2 |
σ* |
0.63019 |
6.15 |
1.54 |
0.087 |
|
N 1 – C 8 |
σ |
1.97294 |
N 1 – C 6 |
σ* |
0.25885 |
3.07 |
1.17 |
0.054 |
|
N 1 – C 8 |
σ |
1.97294 |
C 2 – C 3 |
σ* |
0.2903 |
1.26 |
0.94 |
0.034 |
|
N 1 – C 8 |
σ |
1.97294 |
C 2 – C 9 |
σ* |
0.6117 |
1.61 |
1.52 |
0.045 |
|
N 1 – C 8 |
σ |
1.97294 |
S 5 – C 6 |
σ* |
0.12725 |
1.08 |
1.03 |
0.03 |
|
S 25 – C 29 |
π |
1.96881 |
C 18 – C 28 |
π ∗ |
0.5923 |
13.24 |
1.51 |
0.127 |
|
C 26 – N 27 |
π |
1.84051 |
C 28 – C 29 |
π ∗ |
0.38277 |
12.17 |
0.43 |
0.112 |
|
N 27 – C 28 |
π |
1.96909 |
C 26 – N 30 |
π ∗ |
0.3879 |
10.38 |
1.5 |
0.112 |
|
C 28 – C 29 |
π |
1.96017 |
C 18 – C 28 |
π ∗ |
0.5923 |
10.06 |
1.51 |
0.11 |
|
C 28 – C 29 |
π |
1.8073 |
C 18 – N 19 |
π* |
0.3175 |
24.86 |
0.33 |
0.083 |
|
C 26 – N 27 |
0.55593 |
11.32 |
0.33 |
0.06 |
||||
|
N 1 |
LP (1) |
1.72425 |
C 2 – C 3 |
σ ∗ |
0.64849 |
11.21 |
0.99 |
0.1 |
|
S 5 |
LP(2) |
1.7487 |
C 2 – C 3 |
σ ∗ |
0.36511 |
19.37 |
0.28 |
0.068 |
|
O 10 |
LP(1) |
1.95441 |
C 9 |
σ* |
0.85946 |
12.39 |
1.48 |
0.122 |
Table 4: Natural bond orbital analysis of 6CA2C.
Drug likeness
An active pharmaceutical drug can be studied based on Lipinski rule of five conditions, through which drug likeness parameters are identified and compared [29,30]. Drug parameters with their respective range obtained through Lipinski rule of five were tabulated in Table 5. All these values of drug likeness parameters fall in the desirable range; hence the title compound can be applicable as a pharmaceutically potential candidate.
| Descriptor | Values | Desirable range |
| Hydrogen bond donor (HBD) | 4 | <5 |
| Hydrogen bond acceptor (HBA) | 9 | <10 |
| A logP | 0.92 | <5 |
| Polar surface area (PSA) [Å2] | 109.91 | <140 |
| Molar refractivity | 93.59 | 40-130 |
| Number of rotatable bonds | 9 | <10 |
Table 5: Calculated Drug likeness parameters for 6CA2C.
Molecular docking studies
Primary proteins such as antibacterial protein (4AAW) [31], a DNA ligase inhibitor protein (4GLW) [32] and Peptidoglycan receptor (2F2L) [33] were used to create docking with ligand 6CA2C. 4AAW antibacterial protein has observed with -2.27 kcal/mol binding energy and bond length 2.77 Ǻ formed in Chain A. 4GLW show a slight high binding energy -3.44 kcal/mol compared to 4AAW formed in Chain B. 2F2L, a peptidoglycan receptor has observed with the maximum binding energy -6.84 kcal/mol compared with other two proteins. It is attached to Chain A forming a bond length of 3.48 Ǻ. This affirms that the titled compound 6CA2C can be considered as a potential anti-bacterial. Primary ligand with antibacterial protein (4AAW), DNA synthesis inhibitor (4GLW) and Peptidoglycan receptor (2F2L) interactions and bond formation are represented in Figures 7 (a) – (c) respectively. Ramachandran plot [34] depicts that most of the amino acids are in the favorable zone. Ligplot version 4.5.3 helps in understanding the two-dimensional interaction profiles of selected protein structures Figure 8. Figures 9 (a) – (c) depicts 2D interaction profile pictures mediated by hydrogen bonds and hydrophobic contacts. The maximum binding energy results show that the title compound can be considered as a potential antibacterial compound.
|
Protein (PDB ID) |
Chain |
Bonded residues |
Bond distance (A) |
Inhibition constant (µm) |
Binding energy (kcal/mol) |
Intermolecular energy (kcal/mol) |
Reference RMSD(A) |
|
4AAW |
A |
LYS389:HZ2 LYS350 LYS391 |
2.77 |
21.74 |
-2.27 |
-5.55 |
75.72 |
|
4GLW |
B |
LYS62:HZ3 THR56 THR44 GLU43 GLY61 VAL40 PRO47 ARG145 |
2.63 |
3.01 |
-3.44 |
-6.72 |
46.40 |
|
2F2L |
A |
ASP354:H3 VAL363 ASN427 GLN393:HE22 PHE335:HZ2 SER391 THR392 |
3.48 |
9.71 |
-6.84 |
-10.12 |
26.33 |
Table 6: Molecular docking parameters of 6CA2C Ligand with 4AAW, 4GLW and 2F2L proteins.
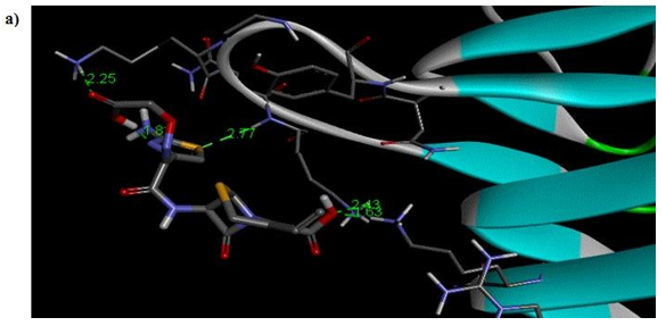
Figure 7a: Molecular docking site with 4AAW receptor.
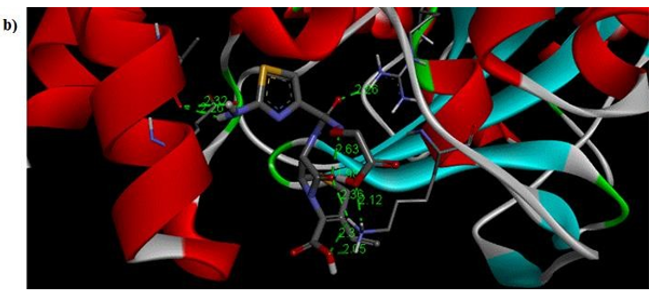
Figure 7b: Molecular docking site with 4GLW receptor.
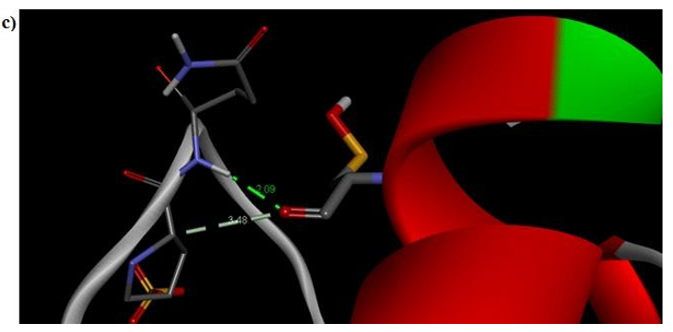
Figure 7c: Molecular docking site with 2F2L receptor.
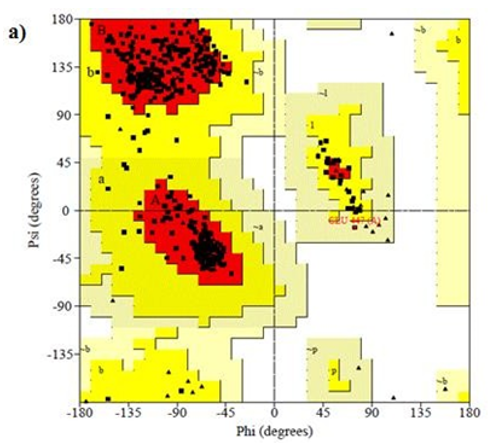
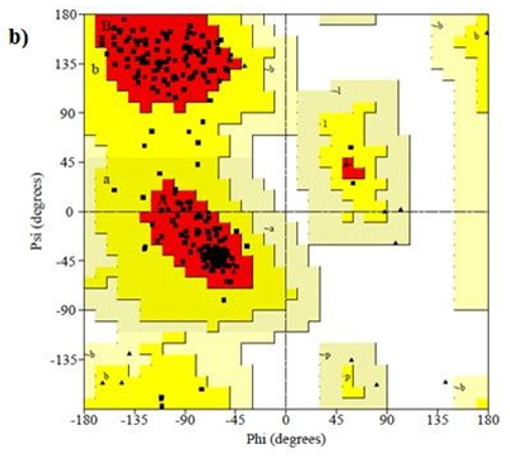
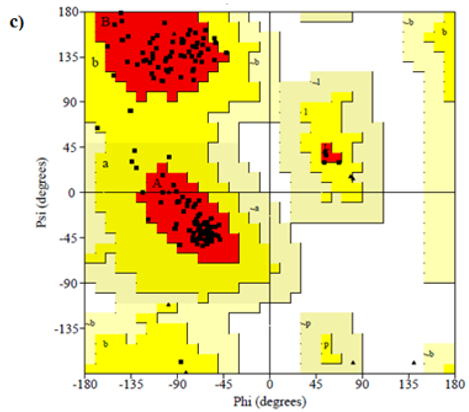
Figure 8: Ramachandran plot of 4AAW,4GLW and 2F2L receptor
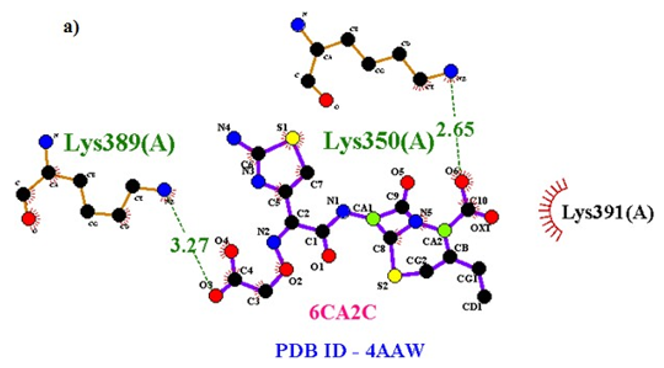
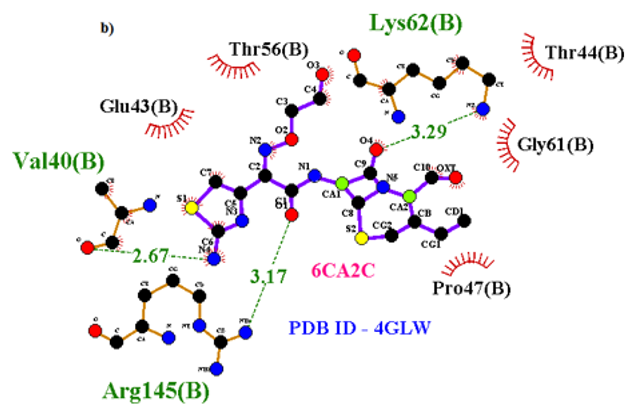
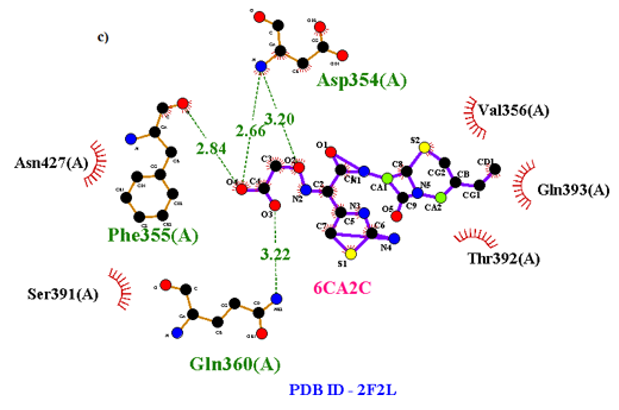
Figure 9: 2D interaction profile representation of 4AAW,4GLW and 2F2L structure
Conclusion
6CA2C has been thoroughly studied in the aspect of theory and experiments. Obtained experimental results were in good agreement with theoretical calculations. This compound was found to be a potential antibacterial drug. In electron-hole analysis, excited state S3 has larger charge transfer length compared to S1 and S2 states. Electronic properties are compared with UV spectral analysis and found both experiments and DFT show concurrence. First order hyperpolarizability (4.36×10-30e.s.u) show larger values identify the molecule as a perfect NLO material. Drug likeness parameters affirm the drug nature of the molecule-based on Lipinski’s rule of five. An active site docking has been made with two proteins such as antibacterial 4AAW and DNA synthesis inhibitor 4GLW show favourable binding with the ligand and therefore the title compound can be considered as an active antibacterial drug. From the above results, it is predicted that the title compound can be effectively used as an active antibacterial compound. However, the experimental analyses and clinical trials must be performed to further confirm the biological activity of the compound.
Funding: No funding from any institution or agency.
References
- Manjusha P, Prasana JC, Muthu S (2018) Quantum mechanical calculations and spectroscopic investigation (FTIR, FT-Raman and UV-Visible) on (6R, 7R)-7-[(2Z)-2-(2-amino-1, 3-thiazol-4-yl)-2-[(carboxymethoxy) imino] acetamido]-3-ethenyl-8- oxo-5-thia-1-azabicyclo [4.2.0] oct-2-ene-2-carboxylic acid: a Pharmaceutical drug using Density functional theory, International Journal of Advanced Research and Development 3: 44-51.
- Cooper RJ, Hoffman JR, Bartlett JG, Besser RE, Gonzales R, et al. (2001) Principles of appropriate antibiotic use for acute pharyngitis in adults: background. Ann Intern Med 134: 509-17.
- Knoller J, Schonfeld W, Bremm KD (1986) In vitro stability of cefixime (FK-027) in serum, urine and buffer. J Chromatogr 389: 312-6.
- Namiki Y, Tanabe T, Kobayashi T, Tanabe J, et al. (1987) Degradation kinetics and mechanisms of a new cephalosporin, cefixime, in aqueous solution. J Pharm Sci 76: 208-14.
- Kumar A, Kelly KJ (1988) In vitro activity of cefixime (CL284635) and other antimicrobial agents against Haemophilus isolates from pediatric patients. Chemotherapy 34: 30.
- Smith SM, Eng RH (1988) Activity of cefixime (FK 027) for resistant gram-negative bacilli. Chemotherapy 34: 455-61.
- Powell M, Williams JD (1987) In vitro susceptibility of Haemophilus influenzae to cefixime. Antimicrob Agents Chemother 31: 1841-2.
- Bowie WR, Shaw CE, Chan DGW, Boyd J, Black WA (1986) In vitro activity of difloxacin hydrochloride (A-56619), A-56620, and cefixime (CL 284,635; FK 027) against selected genital pathogens. Antimicrob Agents Chemother 30: 590-3.
- Noel GJ, Teele DW (1986) In vitro activities of selected new and long-lasting cephalosporins against Pasteurella multocida. Antimicrob Agents Chemother 29: 344-5.
- Neu HC, Chin NX, Labthavikul P (1984) Comparative in vitro activity and β-lactamase stability of FR 17027, a new orally active cephalosporin. Antimicrob Agents Chemother 26: 174-80.
- Kamimura T, Kojo H, Matsumoto Y, Y Mine, S Goto, et al. (1984) In vitro and in vivo antibacterial properties of FK 027, a new orally active cephem antibiotic. Antimicrob Agents Chemother. 25 (1984) 98-104.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JP, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C Pople JA, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009.
- Schlegel HB (1982) Optimization of equilibrium geometries and transition structures. J.Comput.Chem 3: 214-218.
- Parr RG, Yang W (1989) Density Functional Theory of Atoms and Molecules, Oxford University Press, New York.
- Lee C, Yang W, Robert Parr G (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys.Rev 37: 785-789.
- Lehtonen JM, Parkkila S, Vullo D, Casini A, Scozzafava A, et al. (2004) Carbonic anhydrase inhibitors, Inhibition of cytosolic isozyme XIII with aromatic and heterocyclic sulfonamides: a novel target for the drug design. Bioorg.Med.Chem.Lett 14: 3757-62.
- Foresman JB, Frisch AE (2015) Exploring Chemistry with Electronic Structure Methods, 3rd ed., Gaussian, Inc. Wallingford, CT.
- O’Boyle NM, Tenderholt AL, Langner K M (2008) cclib: a library for package-independent computational chemistry algorithms. J. Comp. Chem 29: 839.
- Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J. Comput.Chem 33: 580.
- Muthu S, Ramachandran G, Uma Maheswari J (2012) Vibrational spectroscopic investigation on the structure of 2-ethylpyridine-4-carbothioamide. Spectrochim. Acta A 93: 214-222.
- Muthu SJ, Maheswari U (2012) Quantum mechanical study and spectroscopic (FT-IR, FT-Raman, 13C, 1H, UV) study, first order hyperpolarizability, NBO analysis, HOMO and LUMO analysis of 4-[(4-aminobenzene) sulfonyl] aniline by ab initio HF and density functional method. Spectrochim. Acta A. 92: 154-163.
- Muthu S, Prabhakaran A (2014) Vibrational spectroscopic study and NBO analysis on tranexamic acid using DFT method. Spectrochim. Acta A 129: 184-192.
- Manjusha P, Prasana JC, Muthu S (2018) Quantum mechanical calculations and spectroscopic investigation (FTIR, FT-Raman and UV-Visible) on (6R, 7R)-7-[(2Z)-2-(2-amino-1, 3-thiazol-4-yl)-2-[(carboxymethoxy) imino] acetamido]-3-ethenyl-8- oxo-5-thia-1-azabicyclo [4.2.0] oct-2-ene-2-carboxylic acid: a Pharmaceutical drug using Density functional theory, International Journal of Advanced Research and Development 3: 44-51.
- Diki NYS, Gbassi GK, Ouedraogo A, Berte M, Trokourey A (2018) Aluminum corrosion inhibition by cefixime drug: experimental and DFT studies, J. Electrochem. Sci. Eng. 8: 303-320.
- Christiansen O, Gauss J, Stanton JF (1999) The electronic spectrum of pyrrole, Chem.Phys. Lett. 305 147-155.
- Silvi B, Savin A (1994) Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371: 683–686.
- Heiko J (2008) Localized-orbital locator (LOL) profiles of chemical bonding. Can. J. Chem 86: 695–702.
- Cassidy C, Halbout JM, Donaldson W, Tang CL (1979) Nonlinear optical properties of urea, Opt. Commun 29: 243–247.
- Lipinski CA (2004) Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol 1: 337–341
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23: 3–25.
- Green OM, McKenzie AR, Shapiro AB, Otterbein L. Ni H, et al. (2012) Inhibitors of Acetyltransferase Domain of N- Acetylglucosamine-1-Phosphate-Uridyltransferase/Glucosamine-1-Phosphate- Acetyltransferase (Glmu). Part 1: Hit to Lead Evaluation of a Novel Arylsulfonamide Series. Bioorg Med Chem Lett 22: 1510.
- Surivet JP, Lange R, Hubschwerlen C, Keck W, Specklin JL (2012) Structure-guided design, synthesis and biological evaluation of novel DNA ligase inhibitors with in vitro and in vivo anti-staphylococcal activity. Bioorg Med Chem Lett 22: 6705-6711
- Chang CI, Chelliah Y, Borek D, Mengin-Lecreulx D, Deisenhofer J (2006) Structure of tracheal cytotoxin in complex with a heterodimeric pattern-recognition receptor. Science 311: 1761-1764.
- Laskowski RA, Macarthur MW, Moss DS, Thornton J (1993) PROCHECK: a program to ceck the stereochemical quality of protein structures. J. Appl. Crystallogr 26: 283–291.